Las malformaciones que se producen a nivel de los
tejidos se
denominan hamartomas y se expresan como lesiones
de carácter seudotumoral. La mayor parte de
las veces existe relación entre la malformación y
la alteración de la función,
es decir, las malformaciones se acompañan de una
función alterada con grados variables de
incapacidad funcional. Sin embargo, existen alteraciones
genéticas que comprometen predominantemente la
función de genes, enzimas y que no
necesariamente determinan cambios morfológicos, ya que no
alteran el normal desarrollo de
las estructuras y
corresponden a errores del metabolismo;
en cambio,
aquellas alteraciones de genes que participan en el desarrollo
cuya consecuencia podría manifestarse tardíamente,
deben considerarse entre las malformaciones
A continuación se describirán
anomalías enfocándonos en la parte
craneal.
III.
MALFORMACIONES CRANEALES
Éstas anomalías abarcan desde defectos
mayores incompatibles con la vida hasta aquellos menores e
insignificantes.
Entre estas anomalías tenemos:
a.Acrania.
Esta anomalía la bóveda craneal
está ausente y con frecuencia aparecen anomalías
extensas de la columna vertebral. La acrania asociada a
meroamencefalia o a anencefalia (ausencia parcial del
encéfalo). Ocurre en, aproximadamente, 1 de cada 1000
nacimientos y es incompatible con la vida. La meroanencefalia se
debe a la falta de cierre del extremo craneal del tubo neural a
lo largo de la 4ta semana de desarrollo. Esta anomalía
origina la posterior ausencia de formación de la
bóveda craneal.

b.
Craneocinóstosis.
Se define la craneosinóstosis como el cierre
precoz de una sutura y se utiliza la palabra craneoestenosis
cuando existe una disminución de alguno de los
diámetros craneales.
Se desconoce la causa de la craneosinóstosis. Se
han implicado mutaciones en los genes homeocaja MSX2 y ALX4 en
casos de craneosinóstosis y de otras anomalías
craneales.
b.1
Fisiopatología
– La cabeza del
bebé al nacer está formada por varios huesos que no
están soldados entre sí, sino unidos por unas zonas
más blandas que se denominan suturas. Es en estas zonas
donde se va produciendo el crecimiento del hueso, de manera que
se permita el proceso de
aumento progresivo y homogéneo de la cabeza.
– Las suturas
más importantes son la sagital (arriba, en línea
media, entre los huesos parietales), la coronal (entre el hueso
frontal y los huesos parietales) y la lambdoidea (entre los
huesos parietales y el occipital).
|
| Esquema de las suturas craneales |

– Las zonas de
confluencia de varios huesos y suturas se denominan fontanelas,
que se aprecian al tacto como depresiones blandas, en las que
falta el hueso. La fontanela posterior se forma en la
unión de los huesos parietales con el occipital. Se cierra
alrededor de los cuatro meses de edad. La fontanela anterior se
forma en la unión del hueso frontal con los huesos
parietales y se cierra a partir del año de
vida.
– Al no estar las
suturas cerradas o soldadas, el crecimiento del cerebro es el que
condiciona la forma y tamaño que va a tener la cabeza.
Pero si hay alguna sutura cerrada precozmente, antes del tiempo que le
corresponde, el desarrollo del cerebro va a obligar a los otros
huesos a crecer de una forma anómala para compensar este
fallo. El resultado es el crecimiento de la cabeza de una manera
no armoniosa o simétrica. Este defecto, aunque en
principio es sólo estético, es algo que el
niño arrastrará toda su vida.
– Si todas las suturas
importantes se cierran precozmente, se produce una
situación grave, puesto que no puede el cerebro crecer y
desarrollarse de manera adecuada. De manera que hay un alto
riesgo de que
se produzca retraso psicomotor además de importantes
deformidades craneales y faciales.
– La incidencia global
de estas malformaciones es de un caso de cada 2000 nacimientos.
Con mayor frecuencia afecta a los niños
que a las niñas.
– Algunas de estas
malformaciones son hereditarias y se asocian con otras
malformaciones; pero lo más frecuente es que sean
esporádicas y no hereditarias.
– La gravedad de las
malformaciones craneofaciales dependerá: del número
de suturas cerradas (a mayor número de suturas afectadas,
mayor deformidad y mayor riesgo de compresión cerebral) y
de la presencia o no de anomalías cerebrales
concomitantes.
– Muchas son evidentes
al nacer, pero en algunos casos no se detectan hasta pasados unos
meses. La forma mejor para detectarlas precozmente es
observar:
1. El crecimiento progresivo armonioso y simétrico de
la cabeza.
2. La existencia de las fontanelas y cómo se van
cerrando de manera adecuada a su tiempo.
3. En la palpación de la cabeza del niño
se pueden detectar prominencias óseas a lo largo de una
sutura y esto podría indicar una fusión
precoz de esta sutura.
4. La evolución del perímetro craneal para
detectar si existe un crecimiento inferior a lo normal, al
compararlo con las tablas de crecimiento craneal que existen al
efecto.
b.2
Clasificación
Estas malformaciones se clasifican dependiendo de la
sutura afectada y del número de suturas
implicadas.
· Escafocefalia.- Es la
craneosinóstosis más frecuente. Se produce por el
cierre de la sutura sagital, que une ambos huesos parietales y va
de delante hacia atrás, desde la fontanela anterior a la
posterior. Al no poder crecer
la cabeza a lo ancho, la cabeza se alarga en el sentido
anteroposterior y la región frontal y occipital se abomban
para compensar. La cabeza está alargada y el eje
transversal está disminuido, de ahí que adopte
también el nombre de dolicocefalia (cabeza alargada). Se
suele palpar una cresta ósea a lo largo de la sutura, lo
que da el aspecto a la cabeza de una quilla de barco
(escafocefalia). No suelen presentar síntomas
neurológicos, puesto que el volumen
intracraneal necesario es compensado por el crecimiento del resto
de los huesos y suturas.
|
| Esquema de las suturas en una escafocefalia |

·
|
|
| RX y TAC tridimensional de lactante con escafocefalia | |


· Braquicefalia.- Se produce por
el cierre de toda la sutura coronal, en ambos lados. El
cráneo no puede crecer en el sentido anteroposterior y
queda acortado (braquicefalia). La frente esta aplanada y
ensanchada y las órbitas tienen el aspecto de
“orbitas de arlequín”.
|
| Esquema de braquicefalia |

·
|
| RX de cráneo de una braquicefalia |

· Trigonocefalia.- Se produce
por el cierre de la sutura situada entre los dos huesos
frontales, en la línea media de la frente (sutura
metópica). La frente es estrecha con la forma de la proa
de un barco y se nota una prominencia ósea desde la base
de la nariz hasta la fontanela anterior.
|
| Esquema de trigonocefalia |

·
|
| TAC craneal donde se observa el cierre de la sutura metópica |

· Oxicefalia: se produce por el
cierre de múltiples suturas. La cabeza es muy alargada y
de forma cónica. La turricefalia es una variante en la que
hay un predominio de fusión de la sutura coronal y la
cabeza crece con forma de torre. La oxicefalia y la turricefalia
son situaciones graves en las que no hay compensación de
crecimiento craneal, provocándose una situación de
serio compromiso del desarrollo cerebral.
· Plagiocefalia anterior.- Es
una malformación más compleja en la que se cierra
la sutura coronal en uno de sus lados y de las suturas de los
huesos de la base del cráneo con afectación de la
órbita. Es una craneoestenosis asimétrica, a
diferencia de las anteriores. Se aprecia un aplanamiento de la
frente en un lado con abombamiento del lado contrario. La
órbita afectada está elevada, retraída y
rotada con forma de “órbita de
arlequín”. Se producen deformidades faciales, a
nivel de la nariz y el maxilar superior.
· Plagiocefalia posterior.- El
cierre de la sutura lambdoidea es muy poco frecuente (3 de cada
100.000 nacimientos). En estos casos, se produce un aplanamiento
posterior de uno de los huesos occipitales. También es una
craneoestenosis asimétrica. Es importante diferenciar este
tipo de malformación de la plagiocefalia posicional o
postural del lactante, muy frecuente en los últimos
años y que comentaremos al final de este tema.
La plagiocefalias antes mencionadas serán
más ampliamente descritas en las deformaciones. Esto es
posible ya que los 2 temas no son excluyentes entre
sí.
|
| TAC craneal donde se observa un aplanamiento posterior |

· Cráneo en hoja de
trébol.- Es una malformación craneal
muy severa con afectación de múltiples suturas.
Además se asocia con afectación cerebral
(hidrocefalia), lo que complica aún más la
situación de hipertensión intracraneal.
Existen otras malformaciones muy severas, en las que
predomina la afectación sobre el macizo facial, la
repercusión en la cara del recién
nacido:
· Síndrome de Apert.-
Consiste en la presencia de craneosinóstosis
múltiple, huesos faciales poco desarrollados y
sindactilias (fusión de las falanges de los dedos) en las
manos y/o pies. Se asocia con retraso psicomotor.
· Síndrome de
Crouzon.-Cursa con craneosinóstosis
múltiple, hipoplasia del maxilar y afectación de
órbitas con exoftalmos.
c. Microcefalia.
Los niños aquejados de este trastorno nacen con
una bóveda craneal de tamaño menor o ligeramente
menor. Las fontanelas se cierran durante la etapa temprana de la
lactancia y
las suturas lo hacen durante el primer año. Pero esta
anomalía no se debe al cierre prematuro de las suturas,
sino que es consecuencia del desarrollo anómalo del
sistema nervioso
central (SNC) por lo cual no crece el encéfalo, y por
lo tanto, tampoco el cráneo.
Por lo general los individuos con microcefalia tienen un
retraso mental grave.
c.1 Posibles causas
La microcefalia puede ser provocada por la exposición
a sustancias nocivas durante el desarrollo fetal o quizás
puede estar asociada con problemas o
síndromes genéticos hereditarios.
Las teorías
sugieren que los siguientes factores pueden predisponer al
feto a padecer
los problemas que afectan el desarrollo normal de la cabeza
durante el embarazo:
· Exposición a químicos o
substancias peligrosas.
· Exposición a la radiación.
· Falta de vitaminas y
nutrientes adecuados en la alimentación.
· Infecciones.
· Consumo de alcohol o de
medicamentos recetados o ilegales.
· Diabetes
materna.
La microcefalia puede presentarse como una única
anomalía o en asociación con otros problemas de
salud y puede ser
la consecuencia de la herencia de un
gen autosómico recesivo, o en muy raras
ocasiones, un gen autosómico dominante. El trastorno puede
producirse luego del nacimiento debido a diferentes lesiones
cerebrales.
Autosómico recesivo y autosómico dominante
son dos patrones en los cuales los genes se heredan en una
familia. Los
genes determinan nuestros rasgos como por ejemplo, el color de ojos y
el grupo
sanguíneo, y también pueden provocar una
enfermedad. Autosómico significa que afecta a hombres y a
mujeres por igual, mientras que recesivo significa que, para
padecer la enfermedad (en este caso, la microcefalia), son
necesarias dos copias del gen, una heredada de la madre y otra
del padre. Luego de tener un hijo con microcefalia
autosómica recesiva, los padres tienen un 25 por ciento de
posibilidades (una en cuatro) de tener otro niño con el
mismo trastorno.

Involución con microcefalia
evolutiva y dismorfia.
d. Cráneo
bífido
Los defectos de la formación del cráneo
bífido se acompañan de anomalías
congénitas del encéfalo, meninges o ambos. Estos
defectos del cráneo se encuentran por lo general en el
plano medio y en la bóveda craneal. Con frecuencia, el
defecto se encuentra en la parte escamosa del hueso occipital y
puede incluir la porción posterior del agujero occipital.
Por lo común, cuando el defecto del cráneo es
pequeño, solo se hernian las meninges y el trastorno se
denomina meningocele craneal o cráneo
bífido con meningocele.
El cráneo bífido con herniación de
cerebro, meninges o ambos ocurre en alrededor de 1 de cada 2000
nacimientos. Cuando el defecto craneal es grande, se hernian las
meninges y parte del encéfalo, lo que forma un
meningoencefalocele. Si la parte saliente del
encéfalo contiene una porción del sistema
ventricular, el defecto se denomina
meningohidroencefalocele.
IV.
DEFORMACIONES CRANEALES
a. Plagiocefalia
Posicional.
a.1 Definición.
La plagiocefalia posicional es conocida también
en la literatura por
otros términos como por ejemplo: plagiocefalia por
moldeamiento, plagiocefalia occipital, plagiocefalia deformativa,
plagiocefalia sin craneosinostosis, plagiocefalia postural,
plagiocefalia funcional y plagiocefalia posterior.
En este trabajo la
denominaremos plagiocefalia posicional, porque probablemente es
el que menos confusión genera.
Atendiendo a la etiología de la
deformación, la plagiocefalia posicional es de
“carácter externo” porque está
producida por fuerzas mecánicas externas que actúan
sobre la sutura lambdoidea o la región posterior del
cráneo, bien sea durante la vida intrauterina o
posteriormente, a diferencia de la plagiocefalia
craneosinostótica, que como todas las craneosinostosis es
debida a factores intrínsecos que afectan a las propias
suturas craneales (suturas lambdoideas).
a.2 Causas.
Diversos factores pueden actuar sobre la cabeza fetal
produciendo un fenómeno de moldeamiento craneal:
posiciones fetales prolongadas, embarazos múltiples,
anomalías uterinas (útero bicorne), macrocefalia,
grandes fetos, partos con fórceps o ventosas..
etc.
Después del nacimiento son también muy
numerosas las causas que pueden ocasionar esta deformación
por moldeamiento:
– Una posición elegida por el lactante sin una
razón clara.
– El apoyo sistemático de la cabeza en la
región occipital de un lado o bilateralmente, durante el
sueño o en períodos de despertar.
– La utilización constante de los modernos
“carritos” portadores de los lactantes con el
niño apoyando siempre la cabeza de la misma
forma.
– La presencia de tortícolis debido a muy
diversas etiologías.
– Lesiones en los nervios oculomotores como el IVº
par o de la musculatura ocular.
– Por último numerosas lesiones, incluso
cerebrales, que favorecen la aparición de una
plagiocefalia al disminuir la motilidad espontánea del
niño.
a.3 Incidencia
En la actualidad la incidencia real de la plagiocefalia
posicional es imposible de establecer, pero hay trabajos
recientes que indican que si los criterios diagnósticos
empleados no son correctos, la cifra puede llegar hasta un 48% de
niños sanos por debajo del año de edad.
Por otra parte la plagiocefalia posicional puede ser
unilateral o también bilateral (paquicefalia) en un 20% de
los casos.
La historia natural de esta
deformidad es difícil de establecer claramente24,26, pero
puede suponerse que en la adolescencia o
la edad adulta un número indeterminado de estos
niños pueden presentar alguna deformidad craneal o facial,
generalmente en grados menores24.
a.4 Tipos
a.4.1 Plagiocefalia posicional unilateral
Los datos
clínicos más característicos son debidos al
aplanamiento de la región parieto-occipital que hace que
el peñasco se desplace anteriormente y hacia abajo y al
mismo tiempo la región frontal homolateral se adelanta por
crecimiento compensatorio.
Así pues se producirá:
– Aplanamiento y también alopecia de la
región occipital e incluso parietal.
– Abombamiento de la región occipital
contralateral. Es posible también abombamiento
parietal.
– Pabellón auricular ipsilateral adelantado y
descendido.
– Frontal homolateral abombado.
– Raíz nasal centrada.
– Región maxilar homolateral puede estar
también abombada.
La forma del cráneo es la de un paralelogramo.
La plagiocefalia posicional se puede acompañar
además de otra serie de lesiones: tortícolis
frecuente, hasta en un 41%7 para algunos autores, macrocefalia
moderada (40%) con acúmulos extraaxiales de LCR en un
35%7,17 y muy variadas lesiones cerebrales como hidrocefalia,
hemorragia perinatal, infecciones, espina bífida.. etc.
hasta en un 20% de los casos.
En algunas series también se describe
algún grado de retraso psicomotor hasta en un 19%7 o de
dificultades en el
aprendizaje.
a.4.2 Plagiocefalia posicional
bilateral o Paquicefalia
La cabeza adopta un aspecto braquicefálico y es
conocida también con el nombre de paquicefalia. El
aplanamiento afecta a ambas regiones occipitales, con aumento del
diámetro vertical del cráneo en la región
parietal, acentuándose la protusión del
vértex y existiendo además una disminución
del diámetro antero-posterior craneal. La morfología
facial no se afecta, pero vista de perfil la cabeza en los casos
más graves puede parecer que la región occipital ha
sido “cortada por un hacha”.
La cabeza forma un paralelogramo: la región
occipital deformada está aplanada o incluso
“hundida”. La región occipital contralateral
está abombada. La región parietal unilateral puede
también estar abombada así como también la
región frontal unilateral. El pabellón auricular
puede estar adelantado en el lado de la lesión e incluso
el zigoma también puede protuir moderadamente en el mismo
lado. La raíz nasal suele estar centrada.
a.4.3 Plagiocefalia occipital por
craneosinostosis.
El aplanamiento occipital se ve compensado sobre todo
por abombamiento de la región occipito-mastoidea. La
típica cresta ósea encima de la sutura lambdoidea
suele estar presente y es palpable. Además el abombamiento
contralateral suele ser más parietal que occipital. El
peñasco se desplaza posteriormente, por lo que el
pabellón auricular también se mueve en esa dirección.
Finalmente el cráneo adopta una forma más bien
trapezoidal.
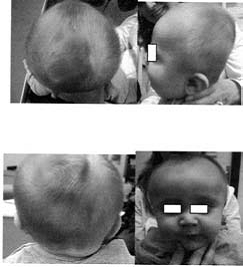
Plagiocefalia posicional
bilateral. La región occipital está aplanada
bilateralmente. Diámetro vertical del cráneo
aumentado. Frente abombada y diámetro biparietal
ensanchado.
b. Trigonocefalea
b.1 Definición
Es una malformación que resulta
del cierre prematuro de la sutura frontal o metópica. La
frente es estrecha y prominente en su linea media con una forma
triangular y se aprecia hipotelorismo. Su interés se
fundamenta en el aspecto estético y su posible
asociación a malformaciones neurológicas
intracraneales.
b.2 Tipos
b.2.1 Simple
Los pacientes con cráneo
trigonocefálico se pueden clasificar en dos grupos: uno, en
el que la sinostosis metópica es un defecto aislado, y
otro grupo en el que las malformaciones importantes y el retraso
mental severo son evidentes.
La trigonocefalia, como síntoma
aislado, es normalmente esporádica. Se ha observado
sólo en las familias con un modo autosómico
dominante y un modo genético recesivo de unión X.
En general, la trigonocefalia simple tiene un buen
pronóstico para la supervivencia y el desarrollo mental,
tanto si se ha empleado la cirugía, como si no.
b.2.2
Compleja
La trigonocefalia compleja abarca un
grupo genético heterogéneo de pacientes. Se ha
observado una variedad de micro-supresión
cromosómica y de síndromes de duplicación,
por ejemplo: supresiones de 3p, 9p y 11q y duplicación de
3q.
Como originalmente lo describió
Opitz, el síndrome de C, trigonocefalia, es un
síndrome de Retraso Mental/Anomalías Mentales
Congénitas, es cierto que no prueba la existencia de un
anomalía cromosómica incluso después de
aplicar las técnicas
de bandas de alta resolución.
Parece que la mayoría de las
características faciales de los pacientes con el
síndrome C de Opitz pueden considerarse como una
<secuencia> trigonocefálica en vez de las
características múltiples de un síndrome
específico. En particular, los pliegues del epicanto, las
hendiduras palpebrales sesgadas hacia la parte superior y el
puente nasal deprimido son el resultado natural de un cierre
prematuro de la parte inferior de la sutura metópica. Los
síntomas faciales más específicos son las
orejas bajas y anormalmente modeladas (100%) y las
anomalías intra-orales: rebordes alveolares amplios que se
estrechan hasta un profundo surco en la línea media del
paladar alto y arqueado y frénula unida.
Las anomalías intra-orales no son
obligatorias pero, si se presentan en combinación con la
trigonocefalia forman el diagnóstico de este síndrome. Las
anomalías en miembros de los pacientes con el
síndrome C, son bastante variables, van desde la
polidactilia postaxial de las manos, y manos y pies acortados, a
la desviación ulnar de los dedos y un ligero acortamiento
rizomélico de las extremidades superiores. Las
anormalidades extracraneales son variables y son relativamente no
específicas, indicando heterogeneidad clínica y
genética.
En muchos de los pacientes el pronóstico es pobre por un
fallo en el crecimiento, anomalías cardiacas, accesos e
hipotonía severa que conduce a dificultades en la
succión e ingestión. Todos los niños que
sobreviven sufren retrasos mentales de forma severa.
Se sugiere que el síndrome de
trigonocefalia C puede ser un síndrome de
micro-supresión citogenética no detectable. Se
informó repetidamente de duplicaciones como de supresiones
del cromosoma 3q en pacientes con un claro fenotipo C, que
convierte este cromosoma en candidato importante a tener en
cuenta para la localización del gen del síndrome C
de Opitz

Infante con
trigonocefalia.
V.
CONCLUSIONES
– Las malformaciones corresponden a alteraciones en la
forma, producto de un
defecto en el desarrollo.
– Las malformaciones craneales abarcan desde defectos
mayores incompatibles con la vida hasta aquellos menores e
insignificantes
– En la acrania hay una ausencia de la bóveda
craneal y tiene una incidencia de 1 de cada 1000 nacimientos, y
es incompatible con la vida.
– La craneosinóstosis se debe cierre precoz de
una sutura.
– La craneosinóstosis se clasifica en
Escafocefalia Braquicefalia Trigonocefalia
Oxicefalia Plagiocefalia anterior Plagiocefalia posterior
Cráneo en hoja de trébol Síndrome de Apert y
Síndrome de Crouzon.
– En la anomalía llamada microcefalia los
niños nacen con una bóveda craneal de tamaño
menor o ligeramente menor al proedio.
– Los defectos de la formación del cráneo
bífido se acompañan de anomalías
congénitas del encéfalo, meninges o
ambos.
– En cuanto a las deformaciones craneales, se dividen en
plagiocefalia posicional y trigonocefalea.
– La plagiocefalia posicional se divide en
Plagiocefalia posicional unilateral
Plagiocefalia posicional bilateral
o Paquicefalia occipital Plagiocefalia por
craneosinostosis.
– La trigonocefalea también se divide en
Simple y
Compleja.
VI.
BIBLIOGRAFÍA
· Lagman y Sadler. EMBRIOLOGÍA
MÉDICA. CON ORIENTACIÓN CLÍNICA. 9na. ed.,
2004.
· Moore, K y Persaud, T: EMBRIOLOGÍA
CLÍNICA. 7ma ed. Mc Graw.- Hill Interamericana. Mexico,
2004.
· Patten, Bradley M. Carlson, Bruce M.
EMBRIOL´GIA BÁSICA DE PATTEN. 1990.
·
Guardia Massó. MEDICINA
INTERNA. 2da ed. Editora Barcelona. Barcelona, 2004.
· Morrison, C.S., Chariker, M.: Positional
plagiocephaly: pathogenesis, diagnosis and management. J Ky
Med Assoc. 2006; 104: 136-140.
· http://centros.uv.es/web/departamentos/D40/data/informacion/E125/PDF385.pdf
·
http://www.neurocirugia.com/diagnostico/trigonocefalia/trigonocefalia.html
· escuela.med.puc.cl/publ/manualcabezacuello/malformaciones.html
· www.sepeap.org/archivos/revisiones/dismorfologia/craneosinostosis.htm
Autor:
Luis Antonio Silva Trujillo
luixtreme_13[arroba]homail.com
UNIVERSIDAD NACIONAL DE TRUJILLO
FACULTAD DE CIENCIAS
MÉDICAS
ESCUELA DE MEDICINA
DEPARTAMENTO DE MORFOLOGÍA HUMANA
ÁREA DE EMBRIOLOGÍA
Promoción: XLVI
Docentes: Dr. Rodil Cruzalegui H.
Dr. Guillermo Fonseca R.
Dr. Javier Álvarez C.
Dr. Luis Florián Zavaleta
TRUJILLO – PERÚ
 Página anterior Página anterior | Volver al principio del trabajo | Página siguiente  |