Ambos se desarrollan a partir del mesodermo. El
mesodermo embrionario adyacente al notocordio en desarrollo (eje embrionario) y
el tubo neural, se engrosan para formar dos columnas
longitudinales llamadas mesodermo paraxial. Estas columnas
mesodérmicas pronto se dividen en segmentos pares que se
denominan somitas. Las somitas forman elevaciones en la
superficie dorsolateral del embrión. Cada somita consiste de
un esclerotoma y un dermomiotoma. Las células
mesenquimatosas dejan los esclerotomas y envuelven el notocordio.
Aquí dan lugar a la columna vertebral y a las costillas. Las
células mesenquimatosas de las regiones del miotomo derecho
dermomiotoma, crean los músculos del dorso. Las regiones del
dermatoma de los dermomiotomas originan la dermis
cutánea.
1. HUESOS
· Desarrollo óseo
Los huesos aparecen al principio como
condensaciones de células mesenquimatosas que constituyen
los modelos o moldes
mesenquimatosos de los huesos. Algunos huesos se desarrollan en
este mesénquima (tejido conectivo embrionario) por
osificación intramembranosa. En otros casos, los modelos
óseos mesenquimatosos se transforman en modelos
cartilaginosos de la siguiente manera: las células
mesenquimatosas que se han agregado en la formación del
futuro hueso, se diferencian en células cartilaginosa
embrionarias llamadas condroblastos. Estas células secretan
matriz cartilaginosa, de
manera que el modelo óseo pronto se
convierte en cartílago hialino. El modelo óseo
cartilaginoso se osifica más tarde por formación
ósea endocondral.
El desarrollo óseo se conoce como osificación
u osteogénesis, hay mecanismos distintos. Todos los huesos
son derivados del mesénquima, pero mediante dos procesos diferentes,
según los huesos involucrados. Por ejemplo, los huesos
planos del cráneo, se desarrollan directamente en áreas
del mesénquima vascularizado por medio de un proceso conocido como
osificación intramembranosa. El proceso se llamó
así porque el sitio de estos huesos aparece al principio
como una membrana mesenquimatosa. Los huesos largos, como se
mencionó más arriba, son precedidos por modelo
cartilaginosos. La mayor parte del cartílago en estos huesos
es reemplazado durante la vida fetal por tejido óseo durante
un proceso conocido como osificación endocondral.

Figura 1-1. Cortes, secciones
transversales de embriones a diversos
estadios
1.1. ESQUELETO AXIAL
1.1.1. Desarrollo de la columna vertebral
Las vértebras se dividen de regiones de los
esclerotomas de los somitas. Las células mesenquimatosas de
estas regiones migran hacia el plano medio y rodean el
notocordio. Cada vértebra forma la condensación de
células mesenquimatosas a partir de la mitad caudal de un
esclerotoma, que fusiona con células mesenquimatosas unidas
de manera laxa, de la mitad craneal del siguiente
esclerotoma.
El notocordio persiste a través de los estadios
mesenquimatosos y cartilaginoso del desarrollo vertebral, pero
tarde o temprano desaparece, entre tanto ocurre la
osificación de las vértebras. El derivado adulto del
notocordio es el núcleo pulposo, el cual forma la parte
central del disco intervertebral.
Mientras continúa el desarrollo, aparecen
apófisis de las vértebras en crecimiento; apófisis
espinosa, arco vertebral, dos apófisis transversas, y dos
costales. La apófisis que forman el arco vertebral (arco
neural), crecen en dirección dorsomedial y se funden una con
otra en el plano medial para encerrar la médula espinal en
desarrollo. Algún trastorno en este proceso para encontrarse
y fusionarse origina un defecto óseo en el arco vertebral
conocido como espina bífida oculta. Si los arcos vertebrales
de varias vértebras son incapaces de desarrollarse en forma
normal, el defecto óseo combinado puede permitir que las
meninges (membranas) y la médula espinal presenten una
hernia, lo que produce una forma grave de espina bífida,
conocida como espina bífida cística (es decir,
meningocele y meningomielocele).
Las apófisis transversas crecen en dirección
lateral a partir de las vértebras y los procesos costales en
dirección ventrodorsal en la pared costal. En la pared
torácica, la apófisis costales forman las
costillas.
Durante o un poco después de la pubertad (de 12 a 16
años), aparecen cinco centros secundarios de
osificación en las vértebras. Todos los centros
secundarios se unen con el resto de las vértebras alrededor
de los 25 años.

Figura 1-2. Ilustraciones de
varios estadios en el desarrollo de una
vértebra.
1.1.2. Desarrollo del cráneo
El cráneo se desarrolla en dos porciones: un
neurocráneo y un viscerocráneo. El neurocráneo se
divide en 1) un neurocráneo
membranoso que da lugar a los huesos planos del
cráneo, por ejemplo, huesos parietales, que rodean el
cerebro y forman la bóveda
del cráneo (cúpula craneal); y 2) un
neurocráneo cartilaginoso o
condrocráneo que forma la base del
cráneo.
Al nacimiento, los huesos planos del cráneo se
separan uno de otro por suturas de tejido conectivo. En
áreas donde se encuentran más de dos huesos, las
suturas son amplias y se conocen como fontanelas. La más
prominente de estas es la fontanela anterior, la cual se localiza
en la unión de los dos huesos parietales con las dos partes
del hueso frontal. Las suturas y fontanelas del cráneo
permiten que los huesos se trasplanten uno sobre el otro durante
el nacimiento, esto permite a la cabeza pasar a través del
conducto del nacimiento (conducto cervical vaginal). Varias de
estas estructuras y fontanelas
permanecen membranosas durante un tiempo considerable
después del nacimiento. Por ejemplo, la fontanela anterior
(llamada a menudo "punto suave" o "mollera”) por lo general
se cierra alrededor de la mitad del segundo año.
El viscerocráneo, forma los huesos de la cara y,
sobre todo, se deriva a partir de los cartílagos de los dos
primeros arcos branquiales.
1.2. ESQUELETO APENDICULAR
1.2.1. Desarrollo del esqueleto
apendicular
El esqueleto apendicular consiste en cintura pectoral
(hombro), cintura pélvica y huesos de las extremidades.
Estos últimos aparecen al principio como condensaciones
mesenquimatosas en la quinta semana. Posteriormente se
desarrollan los centros de condrificación, modelos
cartilaginosos de los huesos que se desarrollarán en la
sexta semana.
· Los centros primarios de
osificación.- Se desarrollan en los huesos
largos y la osificación se inicia hasta el final del periodo
embrionario (56 días). Hacia la decimosegunda semana,
aparecen centros primarios en casi todos lo huesos de las
extremidades.
· Los centros de osificación
secundarios.- Aparecen por lo regular justo antes
del nacimiento de los huesos que forman la articulación de
la rodilla. Sin embargo, la mayor parte de los centros de
osificación secundaria aparecen después del
nacimiento.
El hueso que se forma a partir de un centro primario no
se fusiona con el formato a partir del centro secundario hasta
que el hueso alcanza sus dimensiones de adulto. Conocer las
épocas de aparición de los diversos centros de
osificación, es útil para los radiólogos para
determinar si el esqueleto de un niño crece de manera
normal.

Figura 1-3. Dibujo del cráneo fetal a
las 20 semanas, que indica la derivación de sus
huesos.
II.
ANOMALÍAS CONGÉNITAS
ÓSEAS
Según la clasificación internacional de las enfermedades, ICD 10 tenemos:
ENFERMEDADES CONGENITAS Y MALFORMACIONES
Q00-Q99
MALFORMACIONES DEL SISTEMA | |
Q65 | Deformidades congénitas de la |
Q66 | Deformidades congénitas de los |
Q67 | Deformidades osteomusculares |
Q68 | Otras deformidades osteomusculares |
Q69 | Polidactilia |
Q70 | Sindactilia |
Q71 | Defectos por reducción del miembro |
Q72 | Defectos por reducción del miembro |
Q73 | Defectos por reducción de miembro no |
Q74 | Otras anomalías congénitas del (de |
Q75 | Otras malformaciones congénitas de |
Q76 | Malformaciones congénitas de la |
Q77 | Osteocondrodisplasia con defecto del |
Q78 | Otras osteocondrodisplasias |
Q79 | Malformaciones congénitas del |
De todas las enfermedades presentadas anteriormente
nuestro objeto de estudio se centrara en las más comunes del
periodo fetal y postnatal. Estas serán desarrolladas a
continuación tomando el criterio de desarrollo
céfalo-caudal:
II.1.
MALFORMACIONES CONGÉNITAS DE LOS HUESOS DEL CRÁNEO Y
DE LA CARA
1. CRANEOSINOSTOSIS
1.1. DEFINICIÓN
Craneosinostosis es el cierre prematuro de una (o
varias) suturas craneales. Cuando esto ocurre, el cráneo
deja de crecer en la zona sinostosada y compensatoriamente crece
más en las zonas donde las suturas aún no están
osificadas, para así poder acomodar el crecimiento
del cerebro subyacente. Se manifiesta clínicamente por una
deformidad craneal, de grado variable, según que suturas
estén alteradas. De una forma simple podemos decir que el
aspecto de la cabeza de un paciente con craneosinostosis
presentaría una región plana y otra abollonada. La
craneosinostosis puede ocurrir como un hecho aislado o puede ser
parte de un síndrome con otras malformaciones asociadas. Si
la sinostosis es múltiple y severa, puede haber un
impedimento al crecimiento del cerebro, ocasionando desde
hipertensión endocraneana hasta microcefalia y déficit
intelectual. Si la sinostosis no es múltiple, el crecimiento
cerebral es normal y no hay déficit intelectual.

Figura 1-4
Craneosinostosis
1.2. CAUSAS
La craneosinostosis se presenta en uno de cada 2000
nacidos vivos y afecta a los niños con una frecuencia dos
veces mayor que a las niñas.
Este trastorno suele ser esporádico (ocurre por
azar). En algunas familias, la craneosinostosis se hereda de una
de las siguientes maneras:
· gen autosómico
recesivo:Autosómico
recesivo significa que se necesitan dos copias del gen para que
el trastorno se manifieste, una heredada del padre y otra de la
madre, que son portadores. Los padres portadores tienen un 25 por
ciento (una en cuatro) de probabilidades en cada embarazo de tener un niño
con craneosinostosis. Afecta a ambos sexos en igual
proporción.
· gen autosómico
dominante:Autosómico
dominante significa que se necesita un gen para que el trastorno
se manifieste, y el gen se transmite del padre o la madre al hijo
con un riesgo del 50 por ciento en cada
embarazo. Aquí también ambos sexos se ven afectados en
igual proporción.
La craneosinostosis es una característica de muchos
síndromes congénitos diferentes que tienen una variedad
de patrones de herencia y probabilidades de
repetición, según el síndrome específico
presente. Es importante examinar minuciosamente al niño
así como a los miembros de la familia para buscar
señales de una causa sindrómica (trastorno
genético hereditario) de la craneosinostosis como por
ejemplos, defectos de las extremidades, anomalías del
oído o la oreja o malformaciones
cardiovasculares.
1.3. TIPOS
Existen diversos tipos de craneosinostosis, los cuales
reciben nombres diferentes, según qué sutura, o
suturas, están comprometidas. Entre estos nombres se
encuentran los siguientes:
1.3.1. ESCAFOCEFALIA: El cierre precoz
y exclusivo de la sutura sagital que separa a los huesos
parietales, lleva al crecimiento del cráneo en paralelo a la
sutura cerrada y a la imposibilidad de crecimiento transversal.
El resultado es una cabeza alargada en sentido anteroposterior
(dolicocefalia o escafocefalia) que recuerda a un barco volcado,
correspondiéndose la quilla del mismo con la sutura
fusionada (escafo, es un término griego que significa
barco). No produce hipertensión intracraneal y es, por
tanto, un problema esencialmente estético.

Figura 1-5 Escafocefalia
1.3.2. BRAQUICEFALIA: Cierre precoz de
la sutura coronal que separa a los huesos parietales del
occipital. Si el cierre prematuro se limita a una sutura coronal
el resultado es la plagiocefalia aunque este tipo de deformidad
craneal puede tener otras causas. En el primer caso el
cráneo es transversalmente ancho pero corto en sentido
longitudinal. Puede ocasionar exoftalmos, hipertelorismo,
aplanamiento de la cara e incluso deficiencia mental. En el
segundo, la deformidad es asimétrica con aplanamiento del
lado afectado y de la órbita ocular correspondiente, y
prominencia del lado indemne.
1.3.3. TRIGONOCEFALIA
(cráneo en cuña): Resulta del cierre prematuro
de la sutura frontal o metópica. La frente es estrecha y
prominente y se aprecia hipotelorismo. Su interés es
exclusivamente estético.

Figura 1-6 Trigonocefalia
1.3.4. TURRICEFALIA: Se trata de una
forma mixta, es decir una modalidad de craneosinostosis en la que
se encuentran involucradas varias suturas. Esencialmente el
crecimiento del cráneo es hacia arriba recordando la forma
final al de una torre. Aunque sin acuerdo entre los estudiosos
del tema, suelen describirse dos formas: la Oxicefalia, en la que
el crecimiento es hacia la zona fontanelar, y la
Acrocefalia
cuyo crecimiento es esférico. Ambas originan
retraso mental y trastornos visuales por acodamiento del nervio
óptico.
1.4. SÍNTOMAS
Los bebés con este trastorno presentan cambios en
la forma de la cabeza y la cara que suelen ser evidentes. El
aspecto de la cara del niño puede ser diferente si se la
compara con el otro lado. Otros de los síntomas pueden
incluir los siguientes:
· fontanela abultada (punto blando localizado en la
parte superior de la cabeza)
· somnolencia (o menos alerta de lo normal)
· venas del cuero cabelludo muy
evidentes
· aumento de la irritabilidad
· y llanto fuerte y agudo
· mala alimentación
· vómitos explosivos
· aumento de la circunferencia de la cabeza
· convulsiones
· ojos prominentes e incapacidad del niño de
mirar hacia arriba con la cabeza hacia delante
· retardo del desarrollo
1.5. DIAGNÓSTICO
La craneosinostosis puede ser congénita (estar
presente al nacer) o puede observarse más adelante, durante
un examen físico. El diagnóstico se realiza luego de un
examen físico detallado y las pruebas de
diagnóstico.
Durante el examen, el médico de su hijo
obtendrá los antecedentes prenatales y de nacimiento del
niño y averiguará si existen antecedentes familiares de
craneosinostosis u otras anomalías de la cabeza o del
rostro. Es posible también que el médico le pregunte
acerca de las etapas de desarrollo de su hijo ya que la
craneosinostosis puede asociarse con otros trastornos
neuromusculares. Los retardos del desarrollo a menudo requieren
un seguimiento médico más exhaustivo para poder
así evaluar los problemas
subyacentes.
Durante el examen, se toma una medida de la
circunferencia de la cabeza de su hijo y se representa en una
gráfica para identificar los valores normales y
anormales.
Los exámenes de diagnóstico que pueden realizarse
para confirmar la craneosinostosis incluyen:
· radiografías de la cabeza –
examen de diagnóstico que utiliza rayos de energía
electromagnética invisible para obtener imágenes de los
tejidos internos y los huesos
de la cabeza en una placa radiográfica.
· tomografía computarizada
(También llamada TC o TAC.) – procedimiento de diagnóstico
por imágenes que utiliza una combinación de
radiografías y tecnología computarizada para obtener
imágenes de cortes transversales (a menudo llamadas
"rebanadas") del cuerpo, tanto horizontales como verticales. Una
TC muestra imágenes detalladas
de cualquier parte del cuerpo, incluidos los huesos, los
músculos, el tejido adiposo y los
· órganos. Las tomografías computarizadas
muestran más detalles que las radiografías
generales.
1.6. TRATAMIENTO
Su tratamiento variará según el tipo y los
problemas que acarree. Casi siempre será quirúrgico,
tanto si se buscan mejoras estéticas como si trata de evitar
las graves complicaciones que comportan algunas de las formas de
craneosinostosis. En la actualidad se están investigando
nuevas formas de tratamiento menos agresivas. Si desea más
información al respecto deberá consultar con un
neurocirujano pediátrico que tenga experiencia en este tipo
de enfermedades.
2.
DISOSTOSIS CRANEOFACIAL (SÍNDROME DE
CROUZON)
2.1. DEFINICIÓN
El síndrome de Crouzon es un trastorno
genético. Es uno de muchos defectos congénitos que
provoca la fusión anormal entre los huesos en el cráneo
y rostro. Normalmente, a medida que el cerebro de un niño
crece, las estructuras abiertas entre los huesos permiten que el
cráneo se desarrolle normalmente. Cuando las estructuras se
unen demasiado temprano, el cráneo crece en dirección
de las estructuras abiertas restantes. En el síndrome de
Crouzon, los huesos en el cráneo y rostro se unen demasiado
temprano. Esto provoca una cabeza, rostro, y dientes de forma
anormal. Se cree que la enfermedad de Crouzon afecta a 1 de cada
60,000 personas.

Figura 1.7 síndrome de
Crouzon
2.2. CAUSAS
El síndrome de Crouzon es un trastorno
genético. Es causado por mutaciones (cambios anormales) del
FGFR2 (receptor de factor de crecimiento fibroblasto) o menos
comúnmente de los genes FGFR3. Estos genes ayudan a regular
el desarrollo de las extremidades. Una mutación en estos
genes puede causar que los huesos en el cráneo se unan
demasiado temprano. Investigadores continúan aprendiendo
más sobre las relaciones entre las mutaciones en estos genes
y los varios tipos de síndromes de craneosinostosis que
causan.
2.3. FACTORES DE RIESGO
Un factor de riesgo es aquello que incrementa su
probabilidad de contraer una
enfermedad o condición. Quienes tienen mayor riesgo de
síndrome de Crouzon son hijos de:
§ Padres con el trastorno
§ Padres que no tienen el trastorno, pero que llevan el
gen que causa el trastorno.
§ Padres en edad avanzada al momento de la
concepción
2.4. SÍNTOMAS
Las principales señales y síntomas de síndrome
de Crouzon incluyen:
§ Parte superior y posterior aplastada de la cabeza
§ Frente y sienes aplastadas
§ Parte media del rostro que es pequeña y se
localiza más atrás en el rostro de lo normal
§ Nariz similar a un pico
§ Compresión de pasajes nasales, con
frecuencia causando flujo de aire reducido a través de la
nariz.
§ Mandíbula inferior grande y sobresaliente.
§ Desalineación de los dientes.
§ Paladar estrecho de arco alto, o paladar hendido.
Otros síntomas y complicaciones que pueden resultar por
el síndrome de Crouzon incluyen:
§ Pérdida de la audición
§ Deformidad de los oídos medios
§ Ausencia de canales auditivos
§ Enfermedad de Meniere (mareos, vértigo, o zumbido
en los oídos)
§ Problemas de la visión
§ Ojos cruzados o movimiento ocular
involuntario
§ Curvatura de la columna
§ Dolores de cabeza
§ Articulaciones unidas (en algunos
casos)
§ Acantosis nigrican (porciones pequeñas, oscuras,
aterciopeladas de piel)
2.5. DIAGNÓSTICO
Por lo general, un médico puede diagnosticar
síndrome de Crouzon en el nacimiento o en la niñez
temprana con base en las señales físicas y
síntomas del paciente. Se hacen exámenes para confirmar
el diagnóstico. Estos pueden incluir:
§ Rayos X – una
prueba que usa radiación para tomar una imagen de estructuras internas
del cuerpo, especialmente los huesos
§ Examinación
Genética– exámenes para
confirmar mutaciones en el gen FGFR2 o FGFR 3, los cuales se
pueden usar si los resultados clínicos no son suficientes
para hacer un diagnóstico.
2.6. TRATAMIENTO
No hay cura para el síndrome de Crouzon. Debido a
que no se conoce la causa molecular, científicos están
explorando maneras para bloquear el proceso que conlleva a la
fusión temprana de las estructuras sin afectar otros
procesos importantes de crecimiento. Estos esfuerzos actualmente
se restringen a animales experimentales, pero
avances humanos pueden estar en el horizonte. Actualmente, muchos
de los síntomas se pueden tratar con cirugía.
Además, por lo general se necesita tratamiento
ortodóncico, tratamiento ocular y auditivo, y tratamiento de
apoyo. El buen cuidado dental también es un aspecto
importante para controlar el cuidado de niños con
síndrome de Crouzon.
El tratamiento puede incluir:
§ Cirugía
–Existen numerosas cirugías usadas
para tratar los síntomas del síndrome de Crouzon. Estas
incluyen:
o Craniectomía–
extirpación y reemplazo de porciones del hueso
craneal. Esta cirugía se realiza lo más temprano
posible después del nacimiento para: Prevenir la
presión y daño al cerebro y mantener una forma craneal
que sea lo más normal posible
o Cirugía para tratar exoftalmia
(protuberancia de uno o ambos globos
oculares).Esta cirugía:
– Se realiza directamente sobre la cuenca del ojo o sobre los
huesos que rodean las cuencas de los ojos.
– Puede ayudar a minimizar, pero no eliminar totalmente la
exoftalmia.
2.7. PREVENCIÓN
No se conoce alguna manera para prevenir el
síndrome de Crouzon. Si usted tiene síndrome de Crouzon
o tiene un historial familiar del trastorno, puede hablar con un
asesor genético cuando decida tener hijos.
3.
HIPERTELORISMO
3.1. DEFINICIÓN
El hipertelorismo es un aumento de la distancia de las
paredes internas de la órbita, que en el adulto normal
corresponde a 23-28 mm de distancia interorbitaria (medición
efectuada en una Rx) y de 30-35 mm de distancia intercantal
(medición efectuada en el paciente, tomando como referencia
ambos cantos internos). El hipertelorismo no es en sí un
diagnóstico, sino es la manifestación clínica de
otra patología como podría ser una fisura facial de la
línea media (0-14) o una craneoestenosis o un
tumor.

Figura 1.8 Hipertelorismo
3.2. TRATAMIENTO
El tratamiento es la cirugía ósea con
movilización de las órbitas hacia la línea media,
resecando un segmento óseo en la región de la frente y
la nariz y además tratando la enfermedad de base… Es una
patología de resorte multidisciplinario en que intervienen
el Cirujano Plástico, el Neurocirujano y el
Oftalmólogo.
4. MACROCEFALIA
4.1. DEFINICIÓN
Defecto raro del desarrollo cerebral en el que el
cerebro crece de forma excesiva durante los primeros meses de
vida del niño, a causa de lo cual se produce un crecimiento
anormalmente rápido de la cabeza.
La macrocefalia se determina cuando la medida de la
parte más ancha del cráneo es mayor con respecto a la
medida correspondiente según edad y sexo del paciente.
§ Normalmente la cabeza de un bebé recién
nacido es aproximadamente 2 cm más grande que el tamaño
del pecho o tórax.
§ A los 2 años de edad las medidas son generalmente
casi iguales.
§ Después de los 2 años de edad el pecho
o tórax es más grande que la cabeza.
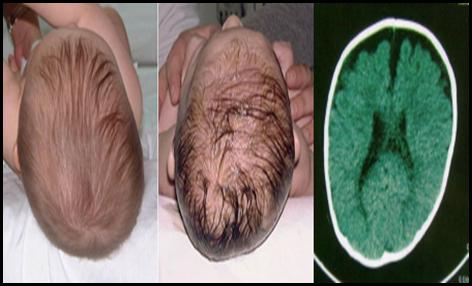
Figura 1.9.
Macrocefalia
4.2. CAUSAS
§ Hidrocefalia (congénita, postraumática u
obstructiva)
§ Enfermedad de Canavan
§ Síndrome de Morquio
§ Macrocefalia familiar benigna
(predisposición familiar al tamaño grande de la
cabeza)
§ Sangrado intracraneal
5. MICROCEFALIA
5.1. DEFINICIÓN
Es el término designado para denominar el
tamaño de la cabeza (distancia alrededor de la parte
superior de la cabeza) significativamente inferior a la media
normal para la edad y el sexo de una persona, sobre la base de tablas
estandarizadas.
La microcefalia usualmente se presenta en la
mayoría de los casos debido a una deficiencia en la tasa de
crecimiento cerebral. El crecimiento del cráneo está
determinado por la expansión cerebral que sucede durante el
crecimiento normal del cerebro en el embarazo y en la infancia.
Las condiciones que afectan el crecimiento cerebral
pueden ocasionar microcefalia, incluyendo infecciones, trastornos
genéticos y desnutrición severa.
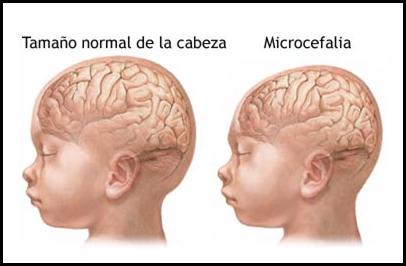
Figura 1.10.
Microcefalia
5.2. CAUSAS
§ síndrome de Down
§ rubéola congénita
§ toxoplasmosis
congénita
§ citomegalovirus congénito
§ síndrome del maullido de gato
§ síndrome de Seckel
§ síndrome de Rubinstein-Taybi
§ trisomía 13
§ trisomía 18
§ síndrome Smith-Lemli-Opitz
§ síndrome de Cornelia de Lange
§ fenilcetonuria (PKU) materna no controlada
§ envenenamiento por metilmercurio
Nota: puede haber otras causas para la
microcefalia, además de las mencionadas. La posibilidad de
incidencia de las mismas no está determinada por el orden en
que éstas se presentan. Entre las causas de este
síntoma se pueden citar enfermedades y medicamentos poco
comunes. Además, las causas pueden variar según la edad
y el sexo de la persona y las características
específicas del síntoma, tales como calidad y enfermedades
asociadas.
6. ANANCEFALIA
6.1. DEFINICIÓN
Es una malformación congénita en la que falta
el encéfalo o tiene un desarrollo rudimentario.
El tubo neural es de las primeras estructuras del
embrión, junto con el corazón; inicialmente es un plato
plano que, cuando el embrión se repliega y crece adelante y
hacia atrás queda finalmente cerrado, como un
tubo.
El tubo neural forma todo lo que son las estructuras de
la columna vertebral, de los nervios, contiene al sistema
nervioso central.
La anencefalia es una malformación grave, porque no
hay desarrollo de hemisferios cerebrales, básicamente hay
rudimentos, o estructuras mínimas de lo que sería el
cerebro.

Figura 1.11. Anancefalia
6.2. CAUSAS
Es una enfermedad de herencia poligénica, hay
muchos genes que predisponen, pero también factores de tipo
ambiental están ayudando a que esta malformación
progrese.
Aún no se comprueba que haya un gen que predisponga
a las hispanas, sin embargo; una de las teorías es que hay
mutaciones en algunas de las personas que impiden que el
ácido fólico se metabolice bien, pues aunque lo
consuman en los alimentos, su organismo no lo
aprovecha.
El ácido fólico es un compuesto muy importante
en la síntesis del DNA, Ácido Desoxirribonucléico,
que es el que da la herencia para todo, la participación del
ácido fólico está en relación directa con la
formación de DNA, si no contamos con lo necesario para
poderlo metabolizar entonces se presentan problemas, de ese tipo,
relacionados con tubo neural, o de algunos otros.
Sí hay una predisposición genética porque
no todas las mujeres tienen embarazos así, o sea vemos que
hay poblaciones con mayor riesgo que otras, entonces en niveles
más elevados económicamente no se ve mucho esto. No
quiere decir que esas parejas no tengan predisposición, pero
su nivel de alimentación es diferente, por eso se piensa
mucho que tienen que ver con la nutrición.
Otros factores de riesgo que predisponen al producto para que desarrolle
esta malformación son:
· La diabetes de la madre puede
provocar la anencefalia en el gestante.
· La presencia de algún cuadro que
provocó fiebre al comienzo del
embarazo.
· La costumbre de algunas personas de ir a los
saunas, los baños de temascal, estando embarazadas sobre
todo en el primer trimestre.
· La hipertermia, el aumento de la temperatura, tanto interna
como externa de la mujer, todo esto en los
primeros meses de embarazo.
No es nada más el que no consuma ácido
fólico, o el que padezca fiebre en los primeros meses,
tienen que ver la predisposición genética o familiar
para que se presenten este tipo de defectos, pero también
pueden desarrollarse por factores externos como sustancias muy
tóxicas, en el agua o en el
aire.
6.3. SÍNTOMAS
A continuación se enumeran los síntomas
más comunes de la anencefalia. Sin embargo, cada niño
puede experimentarlos de una forma diferente. Los síntomas
pueden incluir:
· la parte posterior del cráneo aparece sin
cerrar
· ausencia de huesos en la regiones laterales y
anterior de la cabeza
· plegamiento de las orejas
· paladar hendido – trastorno que se presenta
cuando el techo de la boca del niño no se cierra
completamente, sino que deja una abertura que puede extenderse
hasta la cavidad nasal.
· defectos cardíacos congénitos
· algunos reflejos básicos, pero sin el
cerebro no puede haber consciencia y el bebé no logra
sobrevivir
Los síntomas de la anencefalia pueden parecerse a
los de otros trastornos o problemas médicos. Siempre
consulte al médico de su hijo para obtener un
diagnóstico.
6.4. DIAGNÓSTICO
El diagnóstico de la anencefalia puede realizarse
durante el embarazo o mediante el examen físico del
recién nacido. La cabeza del bebé presenta un aspecto
aplanado debido al desarrollo anormal del encéfalo y a la
ausencia de los huesos del cráneo.
La anencefalia específicamente se puede detectar
desde las 8 semanas, o sea los 2 meses y más adelante
después de las 12 semanas, lo que equivale a los primeros 3
meses.
Los exámenes de diagnóstico que se realizan
durante el embarazo para detectar a los bebés con
anencefalia incluyen los siguientes:
6.4.1. alfafetoproteína –
proteína producida por el feto que se excreta al
líquido amniótico. Los niveles anormales de
alfafetoproteína pueden indicar la presencia de defectos
encefálicos o de la médula espinal, fetos
múltiples, error en el cálculo de la fecha de parto o trastornos
cromosómicos.
6.4.2. Amniocentesis – examen que se
lleva a cabo para determinar la existencia de trastornos
cromosómicos y genéticos, además de ciertos
defectos congénitos. Consiste en insertar una aguja a
través de la pared abdominal y uterina hasta al saco
amniótico para tomar una muestra de líquido
amniótico.
6.4.3. Ecografía (También llamada
sonografía.) –
técnica de diagnóstico por imágenes que utiliza
ondas sonoras de alta frecuencia
y una computadora para crear
imágenes de los vasos sanguíneos, los tejidos y los
órganos. Se utiliza para ver el funcionamiento de los
órganos internos y para evaluar el flujo sanguíneo a
través de diversos vasos.
6.4.4. análisis de
sangre
6.5. PREVENCIÓN
Es importante el consumo suficiente de
ácido fólico para las mujeres que puedan quedar
embarazadas.
Hay buena evidencia de que el ácido fólico
puede ayudar a reducir el riesgo de algunas anomalías
congénitas, incluyendo la anencefalia. Las mujeres que
estén en embarazo o que estén planeando embarazarse
deben tomar un suplemento vitamínico con ácido
fólico todos los días. Muchos alimentos ahora vienen
fortificados con ácido fólico para ayudar a prevenir
estos tipos de anomalías congénitas.
El consumo de ácido fólico en cantidad
suficiente puede reducir en un 50% el riesgo de aparición de
anomalías congénitas del tubo neural.
II.2. DEFORMIDADES CONGÉNITAS DE LA CARA Y DE LA
MANDÍBULA
1. MICROSOMÍA
HEMIFACIAL
1.1. DEFINICIÓN
Es una malformación presente al nacimiento, no es
tan rara (1/ 3.000 RN vivos). Se manifiesta por hipoplasia (en
grado variable) de la hemimandíbula y del maxilar, que
además presenta malformación auricular en ese lado. Es
generalmente unilateral, aunque raramente puede
ser bilateral. No se conoce bien su etiología, pero
se supone que se originaría in útero, por una
alteración vascular en la arteria estapedia (arteria
embrionaria del 1er y 2do arco branquial y precursora del sistema
carotídeo).
Antiguamente se la conocía como Síndrome del
Primer y Segundo Arco Branquial, pues todas las estructuras que
derivan de estos arcos, están alteradas en grado
variable.

Figura 1.12. Microsomía
Facial
1.2. TRATAMIENTO
Su tratamiento comprende la reparación de los
tejidos óseos y blandos alterados. El tratamiento se inicia
en la primera infancia con reparación del nervio facial y
elongación del ramo mandibular (distracción ósea).
A los 6 años de edad se efectúa la reconstrucción
auricular (con cartílagos costales) y en la juventud se efectúa
cirugía del maxilar y la mandíbula (osteotomías e
injertos óseos).
2. SÍNDROME DE TREACHER COLLINS
2.1. DEFINICIÓN
También llamado Disostosis Mandibulofacial.
Clínicamente podría tener un aspecto parecido a la
microsomía hemifacial, pero este síndrome es siempre
bilateral y además no se presenta como casos aislados
debidos a un accidente vascular in útero, sino que tiene un
patrón de transmisión genética autonómico
dominante. Según Tessier, este síndrome
correspondería a la combinación de las fisuras faciales
(6,7 y 8) de la región orbito-malar.
Anatómicamente se presenta como una hipoplasia con
ausencia de hueso (fisura) en el zigoma (o malar) y en la
órbita. Además existe una mandíbula con un ramo
hipoplásico, malformación auricular, inclinación
antimongoloide de los párpados y colobomas (detecto en el
borde libre del párpado).

Figura 1.13.
Síndrome de Treacher
Collins
2.2. TRATAMIENTO
El tratamiento de estos pacientes se inicia en la
infancia y comprende la reconstrucción de partes blandas
(orejas y párpados) así como injertos óseos
vascularizados en el área del malar y cirugías en
maxilar y mandíbula.
3. HIPOPLASIA MAXILAR
3.1 DEFINICIÓN
Es un maxilar superior poco desarrollado en el sentido
antero posterior (retrusión maxilar) o en el sentido
vertical (colapso maxilar vertical). La causa más frecuente
de hipoplasia maxilar es la fisura labio palatina, cuya secuela
es un defecto del crecimiento óseo maxilar.

Figura 1.14.
Hipoplasia Maxilar
4. HIPERTROFIA MAXILAR
4.1. DEFINICIÓN
Es un desarrollo exagerado del maxilar superior. Cuando
es en sentido vertical, ocasiona la Sonrisa Gingival, que es la
exposición exagerada de encía. Cuando la hipertrofia es
en sentido antero posterior, ocasiona el perfil convexo con
dientes salientes y mal oclusión Angle II. Se corrige con
cirugía ósea maxilar.

Figura 1.15.
Hipertrofia Maxilar
5. SÍNDROME DE PIERRE ROBIN
5.1. DEFINICIÓN
Es una micrognatia y retrognatia congénita severa,
de carácter esporádico (no hereditaria) que se asocia a
fisura del paladar y a una lengua grande, por lo cual
estos recién nacidos presentan problemas respiratorios por
obstrucción de la vía aérea superior. Ver manejo
general y de problemas respiratorios en la sección de
Fisuras del Labio y Paladar, manejo del recién nacido
fisurado.

Figura 1.16.
Síndrome de Pierre Robin
II. 3.
MALFORMACIONES CONGÉNITAS DE LA COLUMNA
VERTEBRALY TÓRAX ÓSEO
1. TÓRAX EXCAVADO O EN EMBUDO / TÓRAX EN
QUILLA
1.1. DEFINICIÓN
Un niño con el tórax excavado tiene una
depresión en el centro del pecho, que puede parecer bastante
profunda.
Consulta con un médico si la depresión es tan
profunda como para afectar al corazón y los
pulmones.
El tórax excavado puede ser causado por raquitismo
o puede ser transmitido de padres a hijos. En la mayoría de
los casos las causas se desconocen. Con el tórax en quilla,
el esternón y el pecho del niño salen hacia
fuera.
El tórax en quilla se da más en los niños
que en las niñas. Por lo general en las niñas aparece
muy pronto en la vida, y en los niños, más tarde,
según crecen. El tórax en quilla es menos frecuente que
el tórax excavado, y su causa es desconocida.
Ambos tórax, excavado y en quilla, pueden darse en
forma leve o severa, y pueden asociarse a escoliosis, problemas
respiratorios y cardiopatías.
Ambas condiciones pueden causar dificultades para jugar
o hacer ejercicio. Los niños en edad de crecimiento pueden
experimentar dolor en el pecho e hinchazón y dolor de la
zona del esternón.
· El tórax excavado es el que se "mete hacia
dentro".
· El tórax en quilla es el que "sale hacia
fuera".

Figura 1.17.
Tórax Excavado

Figura 1.18.
Tórax en quilla
1.2. TRATAMIENTO
Si un niño presenta cualquiera de las dos
condiciones, debe ser examinado por un médico. Estas
condiciones se pueden diagnosticar con rayos X.
Los casos leves se pueden corregir por si mismos al
crecer el niño.
La cirugía es recomendable si el defecto del
tórax es moderado o severo, o si el niño tiene
dificultad al respirar o dolores en el pecho al hacer ejercicio.
Acude al médico inmediatamente si esto sucede.
2. ESCOLIOSIS
2.1. DEFINICIÓN
La escoliosis es una curvatura de la columna vertebral.
Un hombro puede estar más alto que el otro, o una cadera
más alta que la otra, lo que afecta el caminar y el
sentarse.
Puede ser causada por raquitismo, por un defecto de la
columna o por causas desconocidas. Afecta más a las
niñas que a los niños.
Las personas con escoliosis deben ser tratadas por un
médico. La escoliosis severa puede afectar a la habilidad de
respirar o de caminar con facilidad y puede causar dolor en las
actividades diarias.

Figura 1.19.
Escoliosis
2.2. CAUSAS
Tres causas generales de escoliosis:
· Congénita que suele estar relacionada con un
problema en la formación de las vértebras o costillas
fusionadas durante el desarrollo prenatal.
· Neuromuscular (control muscular deficiente,
debilidad muscular o parálisis debido a enfermedades como
parálisis cerebral, distrofia muscular, espina bífida y
aunque es historia la secuela de una polio)
· Condición idiopática (de causa
desconocida) que aparece en una columna previamente
derecha
2.3. TRATAMIENTO
La escoliosis se trata mejor cuando el cuerpo de una
persona está todavía creciendo y puede responder a los
tratamientos. Estos incluyen la terapia física para mantener
los músculos flexibles y fuertes, calzados con alzas o
corsés. Los casos severos pueden requerir
cirugía
3. CIFOSIS
3.1. DEFINICIÓN
La cifosis es la curvatura de la columna que produce un
arqueamiento de la espalda, llevando a que se presente una
postura jorobada o agachada.
Es una deformidad de la columna que puede resultar de un
trauma, problemas en el desarrollo o una enfermedad degenerativa.
Esta condición puede ocurrir a cualquier edad, aunque es
rara en el momento del nacimiento.
La cifosis adolescente, también conocida como
enfermedad de Scheuermann, puede ser producto de la
separación de varias vértebras (huesos de la columna)
consecutivas y se desconoce la causa.
En los adultos, la cifosis se puede dar como resultado
de fracturas osteoporóticas por compresión,
enfermedades degenerativas como la artritis, o espondilolistesis
(deslizamiento de una vértebra hacia adelante sobre otra
vértebra).

Figura 1.20.
Cifosis
3.2. CAUSAS
Hay otras causas para la
cifosis como:
· Infección (tuberculosis)
· Neurofibromatosis
Trastornos del tejido conectivodistrofia
muscular
· Espina bífida (deformidad congénita con
formación incompleta de una parte de la columna)
· Degeneración de los discos
· Ciertas enfermedades endocrinas
· Enfermedad de Paget
· Polio
· Tumores
La cifosis también se puede presentar asociada con
la escoliosis. Los factores de riesgo de esta condición
están relacionados con sus
causas.
3.3. SÍNTOMAS
Los síntomas más frecuentes en las cifosis son:
· Dolor de espalda
· Fatiga
· Sensibilidad y rigidez en la columna
· Apariencia redondeada de la espalda.
· Dificultad para respirar (en los casos severos)
3.4. TRATAMIENTO
El tratamiento depende de la causa del
trastorno:
• La cifosis congénita requiere una
cirugía correctiva a temprana edad.
• La enfermedad de Scheuermann inicialmente se
trata con un corsé y fisioterapia. De vez en cuando, se
requiere cirugía en caso de curvaturas grandes y dolorosas
(superiores a 60 grados).
• Las fracturas múltiples por compresión
a causa de la osteoporosis se pueden dejar sin
tratar si no se presenta dolor o no se presenta déficit
neurológico, pero es necesario tratar la osteoporosis para
ayudar a prevenir las fracturas futuras. Cuando se presenta una
deformidad o un dolor debilitante se puede optar por la
cirugía.
Es necesario tratar la cifosis secundaria a una
infección o a un tumor de una manera más agresiva, a
menudo, con medicamentos y cirugía.
El tratamiento de otros tipos de cifosis incluye la
identificación de su causa y, en caso de que se presenten
síntomas neurológicos, se puede recomendar la
cirugía.
Los adolescentes que sufren esta
enfermedad tienden a tener un mejor resultado aún si
necesitan cirugía y la enfermedad se detiene una vez que
ellos terminan de crecer. Si la cifosis se debe a enfermedad
degenerativa de las articulaciones o fracturas múltiples por
compresión no es posible la corrección del defecto sin
la cirugía, y el alivio del dolor es menos
confiable.
4. ESPINA BÍFIDA
4.1. DEFINICIÓN
La espina bífida, también conocida como
mielodisplasia, es un trastorno en el cual existe un desarrollo
anormal de los huesos de la columna, de la médula espinal,
del tejido nervioso circundante y del saco con líquido que
rodea a la médula espinal. Este trastorno neurológico
puede provocar que una parte de la médula espinal y de las
estructuras circundantes se desarrollen por fuera y no por dentro
del cuerpo. Dicha anomalía puede producirse en cualquier
parte de la columna vertebral.
4.2. CAUSAS
La espina bífida es un tipo de anomalía
congénita del tubo neural. Estas anomalías, como por
ejemplo, la espina bífida (columna vertebral abierta) y la
anencefalia (cráneo abierto), se presentan en uno de cada
1.000 embarazos.
Durante el embarazo, el cráneo y la columna
vertebral comienzan a desarrollarse en forma de un plato plano de
células que se enrolla para formar un tubo llamado tubo
neural. Si este tubo no se cierra en forma total o parcial y
queda una abertura, se produce lo que se denomina anomalía
congénita del tubo neural abierto (su sigla en inglés
es ONTD). Esta abertura puede quedar expuesta (en el 80 por
ciento de los casos) o puede cubrirse con hueso o piel (en el 20
por ciento de los casos).
La anencefalia y la espina bífida son los tipos
más frecuentes de ONTD, mientras que los casos de
encefalocele (la protrusión de masa encefálica o de su
recubrimiento fuera del cráneo) se producen con mucha menor
frecuencia. La anencefalia ocurre cuando el tubo neural no se
cierra en la base del cráneo; la espina bífida, en
cambio, se produce cuando el
tubo neural no se cierra en algún lugar de la columna
vertebral.
En más del 95 por ciento de los casos, una ONTD se
produce sin que existan antecedentes familiares del trastorno.
Las anomalías tienen su origen en una combinación de
genes heredados de ambos padres que se suma a distintos factores
ambientales. Debido a esto, estas anomalías se consideran
rasgos hereditarios multifactoriales, es decir, "muchos
factores", tanto genéticos como ambientales, que contribuyen
a su aparición.
Algunos de los factores ambientales que contribuyen a
estas anomalías incluyen la diabetes no controlada en la
madre y determinadas prescripciones médicas. Según los
Centros para la Prevención y el Control de las Enfermedades
(Centers for Disease Control and Prevention, CDC), la tasa de
incidencia de las ONTD puede variar de un estado a otro y de un
país a otro.
Las anomalías congénitas del tubo neural
abierto se manifiestan con una frecuencia cinco veces mayor en
las mujeres que en los hombres. Si en una familia existe un individuo con una ONTD, las
posibilidades de que se produzca otro caso se elevan del 3 al 5
por ciento. Es importante tener en cuenta que en el segundo caso
el tipo de anomalía congénita del tubo neural puede ser
distinto. Por ejemplo, un bebé puede nacer con anencefalia
mientras que el segundo puede padecer espina bífida (en
lugar anencefalia).
4.3. TIPOS
Hay tres tipos de espina bífida:
4.3.1. Espina bífida oculta: Es
una apertura en uno o más huesos de la columna vertebral que
no causa daño alguno a la médula espinal.
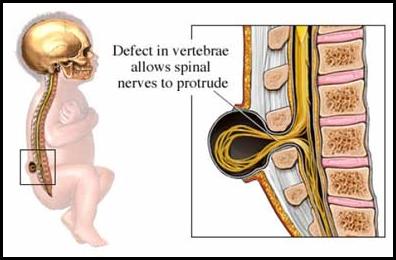
Figura 1.21. Espina bífida
oculta
4.3.2. Meningocele: Es
una condición muy severa de espina bífida en la cual
las meninges que son la cubierta protectora del cordón
espinal escapan al exterior por una apertura en la columna
vertebral. La bolsa que contiene esta parte expuesta al exterior
se conoce como meningocele. La bolsa que puede ser tan
pequeña como una tuerca o tan grande como una toronja puede
corregirse mediante cirugía sin que se le ocasione un
daño significativo a los nervios que componen el cordón
espinal.
Esta bolsa subcutánea contiene principalmente
meninges y fluido, también pueden contener raíces
nerviosas.

Figura 1.22. Meningocele
4.3.3. Mielomeningocele: Es la forma más
severa de espina bífida
. Consiste en una protuberancia de los nervios internos
del cordón espinal a través de una apertura en la
columna vertebral y sin una capa protectora de la piel. El
líquido intrarraquídeo puede gotearse hacia el exterior
y ocasionar un grave problema de infección. Esta
condición suele ocurrir en la parte inferior de la espina
dorsal ocasionando problemas de control de la vejiga e intestinos
del bebé. La lesión espinal puede afectar al tejido
nervioso, meninges y hueso; el saco meningeo contiene una
médula espinal malformada.
Se desarrolla hidrocefalia en más del 80 % de
niños con espina bífida, se produce cuando hay un
aumento del líquido cefalorraquídeo que circula en el
cerebro y alrededor de este, también circula alrededor de la
médula espinal y por el canal central. De existir un bloqueo
en la circulación particularmente en el acueducto o
alrededor de la base del cerebro el líquido
cefalorraquídeo no puede ser absorbido y la presión
comienza a elevarse. En el niño la cabeza puede crecer
fácilmente ya que las suturas del cráneo y las
fontanelas son muy flexibles. La hidrocefalia puede estar
presente en el momento del nacimiento pero usualmente se
desarrolla después del cierre de la lesión espinal,
aumentando rápidamente la circunferencia cefálica. La
hidrocefalia ocurre también en niños que no tienen
espina bífida.

Figura 1.23.
Mielomeningocele
4.4. SÍNTOMAS
A continuación se enumeran los síntomas
más frecuentes de la espina bífida. Sin embargo, cada
bebé puede experimentarlos de una forma diferente. Los
síntomas pueden incluir:
· aspecto anormal de la espalda del bebé, que
puede variar desde una zona pequeña cubierta de vello, un
hoyuelo o una marca de nacimiento hasta una
protrusión en forma de saco ubicada a lo largo de la
columna.
· problemas intestinales y vesicales (por ejemplo,
estreñimiento, incontinencia)
· pérdida de la sensibilidad por debajo de la
zona de la lesión, en especial en los bebés que nacen
con meningocele o mielomeningocele
· incapacidad para mover la parte inferior de las
piernas (parálisis)
El bebé también puede presentar otros
problemas relacionados con la espina bífida, entre los que
se incluyen:
· hidrocefalia (aumento del líquido y de la
presión en la cabeza; se presenta en alrededor de un 80 a un
90 por ciento de los casos)
· problemas cardíacos
· problemas (óseos) ortopédicos
· nivel de inteligencia inferior a lo
normal
Los síntomas de la espina bífida pueden
parecerse a los de otros trastornos o problemas médicos.
Siempre consulte al médico de su bebé para obtener un
diagnóstico.
4.5. DIAGNÓSTICO
Se pueden realizar pruebas de diagnóstico durante
el embarazo para detectar un posible caso de espina fíbida
en el feto. Las pruebas incluyen las siguientes:
4.5.1. Análisis de sangre – El
Colegio Estadounidense de Obstetricia y Ginecología
(American College of Obstetrics and Gynecology, ACOG) recomienda
la sugerencia de un análisis de sangre durante las 15 y las
20 semanas a todas las mujeres embarazadas que nunca tuvieron un
hijo con una ONTD y que no tienen antecedentes familiares de la
anomalía. Este análisis de sangre mide los niveles de
la alfa-fetoproteína (AFP) y otros marcadores
bioquímicos en la sangre de la madre para determinar si su
embarazo corre riesgo de una anomalía congénita del
tubo neural abierto. La AFP es una proteína que normalmente
produce el feto y que atraviesa la placenta y llega a la sangre
de la madre. En general, si un feto padece una ONTD, el nivel de
alfa-fetoproteína en la sangre de la madre será
más elevado. Aunque esta prueba no indica con certeza si un
feto padece una ONTD, determinará qué embarazos corren
mayor riesgo y así realizar pruebas más
específicas.
4.5.2. Ecografía prenatal (También
llamada sonografía.) – técnica de
diagnóstico por imágenes que utiliza ondas sonoras de
alta frecuencia y una computadora para crear imágenes de
vasos sanguíneos, tejidos y órganos. Las
ecografías se utilizan para ver el funcionamiento de los
órganos internos y evaluar el flujo sanguíneo a
través de diversos vasos. La ecografía prenatal puede
detectar una ONTD y se puede utilizar para examinar los
órganos y los sistemas y aparatos del cuerpo
del feto.
4.5.3. Amniocentesis – procedimiento
que consiste en insertar una aguja larga y delgada a través
de la pared abdominal de la madre y dentro del saco
amniótico para extraer una muestra pequeña de
líquido amniótico con el fin de examinarlo. El examen
se realiza para determinar la presencia o la ausencia de una
anomalía congénita del tubo neural abierto. Se debe
tener en cuenta que las anomalías pequeñas o cerradas
pueden no detectarse con esta prueba.
4.6. PREVENCIÓN
Debido a que el tubo neural se cierra entre 28 y 32
días después de la concepción y antes de que
muchas mujeres se percaten de su embarazo, el desarrollo normal
del cerebro y de la médula espinal, durante estas primeras
tres a ocho semanas, puede verse afectado por los siguientes
factores:
· problemas genéticos
· exposición a químicos o substancias
peligrosas
· falta de vitaminas y nutrientes
adecuados en la alimentación
· infección
· medicamentos recetados y consumo de alcohol
A pesar de que se relacionaban muchos factores con el
desarrollo de la espina bífida, las investigaciones demostraron que
el ácido fólico (vitamina B-9), un nutriente
que se encuentra en algunos vegetales de hoja verde, las nueces,
los frijoles, los cítricos y los cereales fortificados para
el desayuno, pueden ayudar a disminuir el riesgo de
aparición de las anomalías congénitas del tubo
neural. Por este motivo, el Colegio Estadounidense de
Genética Médica (American College of Medical Genetics,
ACMG) y los Centros para la Prevención y el Control de las
Enfermedades recomiendan que todas las mujeres tomen un complejo
vitamínico que contenga ácido fólico durante los
años reproductivos. Si una pareja ya ha tenido un niño
con una ONTD, se recomienda la ingesta de una mayor cantidad de
ácido fólico, que puede prescribirla el médico de
la mujer o un profesional de la
salud. Esto permite que la mujer
lo tome durante uno o dos meses antes de la concepción y
durante el primer trimestre del embarazo y así disminuir el
riesgo de gestar otro bebé con ONTD. Los estudios actuales
se centran en analizar cómo los genes controlan la
neurulación o la formación del tubo neural. Comprender
este proceso servirá de ayuda para prevenir defectos del
tubo neural.
 Página anterior Página anterior | Volver al principio del trabajo | Página siguiente  |