INTRODUCCIÓN:
En los últimos años ha aparecido una nueva rama de
la patología denominada crestopatías que estudia las
alteraciones provocadas por modificaciones anormales en células derivadas de la cresta neural.
Las crestopatías tienen importancia en el campo de la
cirugía plástica y maxilofacial porque están
relacionadas con todos los errores en la morfología facial humana.
Como estos defectos se inician en los periodos tempranos de la
embriogénesis dan como resultado malformaciones
congénitas.
MALFORMACIONES CRANEOFACIALES CONGéNITAS Y DEL
DESARROLLO.
Dra. Teresa Pesqueira B.
Pontificia Universidad Católica de
Chile
INTRODUCCIÓN:
Las malformaciones del cráneo y de la cara son un
capítulo amplio y difícil de la cirugía
plástica, que ha evolucionado mucho en los últimos 50
años. De todas las malformaciones que describiremos a
continuación, la más frecuente es la fisura labio
palatina o labio leporino. Esta malformación involucra
sólo la cara y será tratada en un capítulo aparte
debido a su importancia.
Otras malformaciones más complejas y menos frecuentes
alteran tanto los huesos como los tejidos blandos del cráneo y
de la cara, y son las llamadas Malformaciones Craneofaciales.
Las malformaciones craneofaciales , en una forma amplia, se
dividen en aquellas que se relacionan con la aparición de
fisuras (clínicamente corresponde a una hendidura de los
tejidos blandos y de los huesos del esqueleto del cráneo y/o
de la cara.) y en aquellas malformaciones del cráneo y de la
cara que derivan de un cierre prematuro de las suturas craneales,
llamadas sinostosis (clínicamente se manifiestan por
cráneos y caras malformadas producto de un crecimiento
anómalo del esqueleto óseo.)
Algunas veces estas malformaciones craneofaciales complejas,
presentan una concurrencia de hechos que permiten clasificarlas
como síndromes (clínicamente se manifiestan con facies
características y otras anomalías asociadas. Ejemplos
son la enfermedad de Crouzon y el Síndrome de Apert). Por
último, existe un grupo llamado disostosis
craneofaciales (representados clínicamente por el
Síndrome de Treacher Collins y la Microsomía
Hemifacial.)
También forman parte de este capítulo las
malformaciones de la órbita, que revisaremos
brevemente y que también pueden presentarse como un hecho
aislado o formando parte de malformaciones craneofaciales
más complejas. En el enfoque y tratamiento de esta
patología existe una estrecha relación entre los
especialistas en Cirugía Plástica Reconstructiva y los
oftalmólogos.
Por último, las malformaciones del maxilar y la
mandíbula también forman parte de este capítulo y
relacionan íntimamente las especialidades de cirugía
Plástica Reconstructiva y Estética con la
Cirugía Oral y Maxilofacial.
IV. MARCO
TEÓRICO
1) FORMACIÓN DE
LA CARA
Las células de la cresta neural de la zona craneal y
vagal van a dar lugar al ectomesénquima de la región
cráneo-cérvico-facial y arcos branquiales, a partir del
cual se diferencian los procesos faciales.
Una de las características más importantes en la
formación de la cara la constituyen los desplazamientos y
multiplicación celular que dan como resultado la
formación de los mamelones o procesos faciales.
La característica más típica del desarrollo de
la cabeza y cuello es la formación de arcos branquiales o
faríngeos. Aparecen en la cuarta y quinta semana del
desarrollo intrauterino. En un periodo inicial están
constituidos por tejido mesenquimático, separados por surcos
denominados hendiduras faríngeas. Los arcos branquiales no
solo contribuyen a la formación del cuello, sino que
desempeñan un papel importante en la formación de la
cara, principalmente el primer y segundo arcos.
La cara se forma entre las semanas cuarta a octava del periodo
embrionario gracias al desarrollo de cinco mamelones o procesos
faciales: El mamelón cefálico o frontonasal constituye
el borde superior del estomodeo o boca primitiva. Los procesos
maxilares se advierten lateralmente al estomodeo y, en
posición caudal a éste, los procesos mandibulares
(ambos procesos derivados del primer arco branquial).
A cada lado de la prominencia frontonasal se observa un
engrosamiento local del ectodermo superficial, las placodas
nasales u olfatorias. Durante la quinta semana las placodas
nasales se invaginan para formar las fositas nasales. En la sexta
semana aparecen rebordes de tejido que rodean a cada fosita
formando, en el borde externo, los mamelones nasales externos y,
del lado interno, los mamelones nasales internos.
En el curso de las dos semanas siguientes los procesos
maxilares crecen simultáneamente en dirección medial,
comprimiendo los procesos nasales hacia la línea media. En
una etapa ulterior queda cubierta la hendidura que se encuentra
entre el proceso nasal interno y el
maxilar, y ambos procesos se fusionan. En consecuencia, el labio
superior es formado por los dos procesos nasales internos y los
dos procesos maxilares.
El labio inferior y la mandíbula se forman a partir de
los procesos mandibulares, que se fusionan en la línea
media.
La nariz se formará a partir de las cinco prominencias
faciales: la prominencia frontonasal da origen al puente de la
nariz y la frente; los mamelones nasales externos forman las
aletas y los procesos nasales internos fusionados dan lugar a la
punta de la nariz.
Como resultado de la fusión en profundidad de
los procesos nasales internos se forma el segmento intermaxilar,
del cual derivarán el surco subnasal, la porción del
maxilar superior con los cuatro incisivos y el paladar primario
triangular.
Las crestas palatinas derivadas de los procesos maxilares se
fusionan entre sí la séptima semana dando lugar al
paladar secundario. Hacia delante las crestas se fusionan con el
paladar primitivo dejando como línea divisoria entre ambos
paladares el agujero incisivo.
Con la formación de las coanas primitivas se pone en
comunicación las cavidades
nasales primitivas con la cavidad bucal, inmediatamente por
detrás del paladar primario.
El pabellón de la oreja se desarrolla a partir de seis
proliferaciones mesenquimatosas en los extremos dorsales del
primer y segundo arcos branquiales, y rodeando a la primera
hendidura faríngea. Estas prominencias, tres a cada lado del
conducto auditivo externo, se fusionan y se convierten poco a
poco en la oreja definitiva.
Los ojos comienzan a desarrollarse en forma de un par de
vesículas ópticas de cada lado del prosencéfalo al
final de la cuarta semana de la vida intrauterina. Las
vesículas ópticas, evaginaciones del cerebro, toman contacto con el
ectodermo superficial y provocan los cambios necesarios para la
formación del cristalino. La córnea se forma a partir
de ectodermo superficial y epitelio epidérmico. A
través de la fisura coroidea penetra la arteria
hialóidea (futura arteria central de la retina) y las fibras
nerviosas del ojo.
Los párpados aparecen al segundo mes como repliegues
ectodérmicos. Durante el cuarto mes se ponen en contacto y
se sueldan. Al sexto mes se vuelven a separar.
Durante el tercer mes aparecen como finas vellosidades los
pelos de la cara, en primer lugar las cejas y en el labio
superior.
Si bien los arcos branquiales confieren en un periodo inicial
el aspecto típico a la cabeza y el cuello, en el periodo
postnatal la aparición de los cornetes nasales, senos
paranasales y dientes proporcionan a la cara su forma adulta
definitiva.
Tabla 1. Cronología de la formación
de la cara
Cuarta semana | Desarrollo de los arcos branquiales Placodas Aparecen los procesos mandibulares Vesículas ópticas y |
Quinta semana | Fositas nasales Mamelones maxilares superiores Órgano vomeronasal |
Sexta semana | Coanas primitivas Paladar primario Labio superior Esbozos de los senos maxilar y |
Séptima semana | Paladar secundario Conducto auditivo externo y pabellón |
Octava semana | Cara con aspecto humano Esbozos dentarios |
Feto a los tres meses | Fusión palatina completa Cartílago nasal Vello facial |
Cuatro meses | Bullas etmoidales Seno esfenoidal |
Cinco meses | Degeneración del órgano |
Seis meses | Dientes formados Osificación maxilar |
Siete meses | Celdas etmoidales Esbozo seno frontal |
2) MECANISMO DE
SOLDADURA DE LOS MAMELONES
FACIALES
En el periodo embrionario la formación de la cara resulta
del crecimiento desigual de los cinco mamelones o procesos
faciales y de su fusión.
La unión de los distintos mamelones entre sí se
realiza por medio de dos mecanismos: Mesodermización o
Consolidación remodeladora.
·
Mesodermización: Los mamelones se desarrollan de
forma independiente y al alcanzar cierto grado de desarrollo, sus
epitelios se ponen en contacto soldándose sus bordes libres,
siendo posteriormente reabsorbidos por células fagocitarias
mesenquimáticas. De esta manera, los mesénquimas de los
mamelones constituyen uno solo. Este tipo de soldadura tiene
lugar, por ejemplo, en el mecanismo que dará como resultado
el cierre del paladar.
·
Consolidación remodeladora: Los mamelones faciales
son zonas de crecimiento más veloz que las áreas
vecinas, pero en estadios posteriores las áreas que se
desarrollan más lentamente podrán crecer hasta hacer
desaparecer la diferencia superficial que se había
establecido. En la actualidad, se considera que este tipo de
unión es la que realizan la mayor parte de los mamelones
faciales al fusionarse.
Fallos en los procesos de fusión de los diferentes
mamelones desencadenarán la producción de
anomalías en la formación de la cara.
3) MALFORMACIONES
FACIALES
Las causas determinantes de malformaciones faciales pueden
incidir sobre el huevo, el embrión o el feto.
Sobre el huevo inciden factores transmitidos por herencia.
Sobre el embrión y el feto inciden los factores
congénitos, que pueden ser de carácter infeccioso,
mecánico, tóxico o nutritivo.
Las anomalías faciales ocurren por la falta de
coalescencia de los mamelones, por la no mesodermización de
los mismos, o por un desarrollo menor o exagerado de alguna de
sus partes.
Los fallos en la fusión de los mamelones faciales se
pueden explicar porque: uno, o ambos mamelones, no crecen lo
suficiente para contactar con sus homólogos; los mamelones
enfrentados no sueldan sus epitelios o, si hay soldadura, no
existe cohesión necesaria para impedir que más tarde se
separen; que el epitelio sea reabsorbido parcialmente y /o que se
produzca una orientación defectuosa de las áreas de
crecimiento.
Las modificaciones extremas son incompatibles con la vida. En
cambio, las anomalías
pequeñas son frecuentes, al punto que es muy difícil
hallar una cara simétrica por completo.
Las alteraciones craneomaxilofaciales se pueden clasificar en
dos grandes grupos. El primero incluye
síndromes congénitos en los que puede existir
afectación facial como forma de presentación
única, o como un signo más dentro de un complejo
más severo. El segundo grupo incluye trastornos del
crecimiento que aparecen durante el mismo, con causa conocida o
de forma idiopática.
A continuación citaremos algunas de las más
importantes malformaciones congénitas de la cara y del
cuello, ya que de su estudio en profundidad se encargarán
posteriores capítulos.
a) MALFORMACIONES
CONGéNITAS
Cualquier fallo en el desarrollo normal ocasiona
modificaciones en el individuo en gestación.
Conociendo los diferentes mecanismos morfogenéticos de cada
una de las estructuras que forman la cara
podremos determinar la etiopatogenia de sus malformaciones
congénitas.
·
Malformaciones craneales
Las variaciones que presenta el cráneo van desde
imperceptibles hasta aquellas que son incompatibles con la vida.
En la mayor parte de los casos, tanto la forma como el
tamaño del cráneo corresponde a las
características del encéfalo, dado que éste es la
causa de su expansión. Entre las malformaciones craneales se
pueden mencionar:
·
Craneosquisis (hendidura o fisura):
Fisura congénita del cráneo secundaria a un defecto
de la neurulación o cierre de la porción dorsal
segmentaria o no del conducto neural en el encéfalo y de la
subsiguiente inducción de las
cubiertas meníngeas, óseas, musculares y
cutáneas.
·
Acrania:
De acuerdo a la literatura, la secuencia acranea/anencefalia
sola o asociada con otros DTN, son las más frecuentes y la
mayoría pueden estar relacionadas con alteraciones del
metabolismo del AF. Los
defectos cráneo-encefálicos junto a otros defectos
mayores, se asocian a cuadros genético-cromosómicos
(síndromes polimalformativos) y los defectos
cráneo-encefálicos que presentan lesiones por bridas se
relacionan habitualmente con disrupciones por
teratógenos.
Tanto los fetos como los RN se reclasificaron en 4 subtipos de
acuerdo a un patrón fenotípico diferente: A) secuencias
acranea/anencefalias clásicas, aisladas, B)
acranea/anencefalia asociada a otros DTN, C) defectos
cráneo-encefálicos asociados a otras malformaciones
mayores y D) defectos cráneo-encefálicos asociados a
bridas amnióticas ().

·
Craneosinostosis:
Define aquellos procesos secundarios a la obliteración
prematura de una o más suturas del cráneo. Según
la forma resultante del cráneo se clasifican en diferentes
tipos: Escafocefalia, Acrocefalia, Braquiocefalia,
Trigonocefalia, Microcefalia.
Existen craneosinostosis sindrómicas como el
Síndrome Crouzon, el Síndrome de Apert, etc. Dada la
amplitud de este tema, se dedicará un capítulo completo
del libro para su mejor conocimiento.
·
Fisuras faciales
Como consecuencia de la no soldadura de los procesos faciales
entre sí aparecen las fisuras faciales. Se distribuyen
alrededor de la órbita y párpados y de los maxilares y
labios, existiendo fisuras comunes a ambas. Las fisuras de
tejidos blandos y duros no siempre coinciden, y en un mismo
paciente pueden coexistir diferentes fisuras. Se describen hasta
quince tipos.
·
Microsomía hemifacial
Sinónimos: SÍNDROME DE PRIMER Y SEGUNDO
ARCO, DISOSTOSIS OTOMANDIBULAR, DISPLASIA
AURICULOBRANQUIOGÉNICA O SÍNDROME
OROMANDÍBULOAURICULAR
El Síndrome del primer arco branquial corresponde a las
alteraciones que se producen en las regiones derivadas de dicha
zona embriológica. Generalmente varias estructuras son
afectadas simultáneamente.
Entre las teorías existentes para
explicar la lesión, figuran la teoría de la deficiencia
mesodérmica, o defectos en la arteria estapedia.
También la ingesta de talidomida.
La deformidad de la microsomía hemifacial varía en
su extensión y gravedad, pudiendo estar afectadas todas las
estructuras derivadas del primer y segundo arco branquiales.
Básicamente, este síndrome se manifiesta
clínicamente por hipoplasia auricular, maxilar y mandibular,
pudiendo implicar secundariamente a estructuras vecinas: cigoma,
oído medio, nervio facial,
musculatura de la mímica y masticatoria, piel y tejido subcutáneo.
Las malformaciones pueden ser bilaterales, aunque son más
frecuentes las de un solo lado. La ausencia de la rama montante
de la mandíbula provoca secundariamente un desplazamiento
del lado no afectado.
Se han descrito varios síndromes en esta región,
entre ellos: Síndrome de Pierre- Robin (hipoplasia
mandibular, micrognatia), Treacher – Collins (escotaduras
simétricas en parte externa de cada párpado inferior y
deficiente desarrollo de los huesos malares), Disostosis
mandibular, Deformidades de oído interno y medio, Labio y
Paladar Fisurado, Hipertelorismo.
· Fisura
labioalveolopalatina. Labio leporino. Fisura palatina
La Fisura Labioalveolopalatina (FLAP) es la malformación
congénita más frecuente de la región facial.
El agujero incisivo se considera la línea divisoria entre
las deformaciones anteriores y posteriores del paladar.
Las anteriores al agujero incisivo comprenden el Labio
leporino lateral, Fisura del maxilar superior y Hendidura de los
paladares primario y secundario. Estos defectos se deben a la
falta de fusión parcial o completa de los procesos maxilares
superiores con los procesos globulares o extremo inferior de los
mamelones nasales internos, de uno o ambos lados. Aunque se
presenta en pocos casos, es posible la presencia de Labio
Leporino Mediano por escaso desarrollo de los procesos nasales
medianos.
Los defectos situados por detrás del agujero incisivo
dependen de la falta de fusión de las crestas palatinas, y
comprenden la Fisura de paladar secundario, Úvula hendida y
bífida.
En muchas ocasiones, el labio leporino está
acompañado de paladar fisurado.
· Deformidades
dentofaciales
Déficit y excesos mandibulares y maxilares, Mordida
abierta, Sobremordida.
·
Malformaciones branquiales
El complicado desarrollo de la cara a partir de los arcos
faríngeos produce muchas anomalías craneofaciales. De
igual modo, las anomalías de estas estructuras situadas en
la región cervical provocan la formación de quistes
anormales laterales y de la línea media.
Son muy conocidos los Quistes branquiales laterales,
Fístulas y quistes de los senos y de los conductos
tiroglosos, como ejemplos de defectos congénitos en esta
región.
·
Malformaciones nasales
Las malformaciones nasales pueden tener importancia no solo
por la alteración estética, sino también por su
repercusión funcional.
·
Ausencia de nariz, por la no aparición de las placodas
olfatorias.
·
Malformación unilateral; falta de una placoda olfatoria.
·
Nariz bífida; los mamelones nasales internos están
separados, provocando que las narinas estén muy separadas y
que el puente nasal presente un surco muy marcado.
·
Malformaciones del tabique nasal, por falta de coaptación de
los centros de crecimiento.
·
Deformidades de la pirámide nasal: exceso de dorso, nariz en
silla de montar, asimetría nasal, trastornos alares.
·
Malformaciones asociadas a síndromes complejos, como la
fisura labiopalatina.
·
Fisuras y quistes nasales.
·
Atresia de coanas, etc.
·
Malformaciones de la oreja
La oreja y el resto de componentes faciales se originan de los
mismos bloques embriológicos (arcos branquiales mandibular e
hioideo), de ahí que puedan coexistir varias malformaciones
en un mismo paciente.
Dado el diferente origen primitivo del oído interno
(neuroectodérmico) respecto del oído externo y medio
(ectomesénquima), pocas veces el oído interno resulta
comprometido en las alteraciones de la oreja.
Existe gran número de síndromes clínicos con
afectación del oído externo, por ejemplo:
Acondroplasia, Enfermedad de Alport, Enfermedad de Couzon,
Treacher-Collins, etc.
Una de las múltiples clasificaciones de malformaciones
congénitas auriculares es la de Tanzer, la cual reúne
las siguientes anomalías:
·
Anotia
·
Microtia con/sin atresia del conducto auditivo externo
·
Hipoplasia del tercio medio de la oreja
· Oreja
retraída (oreja en copa y caídas), Criptotia,
hipoplasia de todo el tercio superior.
· Oreja
prominente.
·
Asimetrías faciales
En los casos en los que las alteraciones corresponden a un
desarrollo insuficiente de los nervios trigémino y facial,
el resultado se manifiesta en un escaso desarrollo de la
musculatura correspondiente. Cuando la alteración se
presenta de forma unilateral, el lado opuesto puede determinar un
desplazamiento de la línea media hacia el lado normal.
·
Malformaciones de la cara asociadas a otras
anomalías
En algunas ocasiones las anomalías faciales se presentan
asociadas a otras malformaciones que pueden estar ubicadas en
regiones alejadas. Por ejemplo: Necrosis facial unilateral
acompañada de agenesia de un pulmón. El problema
consiste en establecer si ambas malformaciones corresponden a un
mismo factor genético o si se trata de dos alteraciones
simultáneas.
Una vez realizada una somera descripción de algunas de
las malformaciones congénitas craneofaciales,
profundizaremos en los siguientes capítulos en aquellas
consideradas más importantes, tanto por su frecuencia como
por su repercusión estética y funcional.
4) SEGÚN
REFERENCIAS DEL TRABAJO REALIZADO POR LA
DOCTORA TERESA PESQUEIRA B. DE LA PONTIFICIA UNIVERSIDAD
CATÓLICA DE CHILE SE TOMO EN CUENTA LA SIGUIENTE
CLASIFICACIÓN DE LAS ALTERACIONES
CRANEOFACIALES.
Clasificación
Básica
Malformaciones Congénitas del Cráneo y de la
Cara
1.-Fisuras del labio y del paladar (labio leporino)
2.-Malformaciones Cráneo-Faciales
A) Fisuras faciales
B) Cráneosinostosis
Simples
Sindromáticas
C) Disostosis cráneofacial
Microsomía hemifacial
Sindrome de Treacher Collins
3.-Malformaciones de la Orbita:
Hipo e hipertelorismo
Distopía orbitaria
4.-Malformaciones del maxilar y
mandíbula.
FISURAS
DE LABIO Y PALADAR

La fisura del labio y del paladar es una malformación
congénita muy frecuente. En algunos países es la
primera malformación congénita reportada en los
certificados de nacimiento. Su frecuencia es muy alta en algunos
países sudamericanos 1/450 RN vivos.
En USA es de 1/700 RN vivos y en asiáticos de 1/500 RN
vivos. Es un poco menos frecuente en caucásicos y negros.
Tiene carácter genético en un tercio de los casos, pero
en los 2/3 restantes se presenta como un hecho aislado
multifactorial. Si un progenitor tiene labio leporino, el
riesgo de tener descendencia
con la misma patología es de 2%.Si dos padres sanos tienen
un hijo figurado, tienen un riesgo de 5% de tener otro hijo con
esta deformidad.
Hoy en día el tratamiento de esta deformidad es bastante
exitoso. Precisa de un amplio equipo, con la participación
de numerosos especialistas: cirujano plástico,
fonoaudiólogos, terapeutas del lenguaje, ortodoncistas,
otorrinolaringólogos, genetístas, psicólogos,
pediatras, anestesiólogos etc.
Esta patología tan frecuente resulta muy poco conocida
para muchos médicos. A pesar de integrar tantas
especialidades médicas para poder ser tratada
correctamente, no es enseñada a nivel de pregrado, en
ninguna de estas disciplinas médicas.
Anatomía Básica: La nariz normal posee una
columela (porción blanda, central y anterior) recta apoyada
en un tabique central y recto. Los orificios nasales están
formados por los cartílagos alares que nacen como un arco
desde la columela y se apoyan en la base o ala nasal. Ellos son
los responsables de la armonía de la punta nasal.
Inmediatamente caudal se inicia el labio superior, que
presenta un músculo circular, el orbicular, que debe ser
íntegro. En el centro del labio, se sitúa el filtrum
(dos columnas y una depresión central) que
terminan en el arco de cupido (curvo como un corazón). El límite
entre el labio cutáneo y el rojo labial de la mucosa,
está dado por la línea blanca.
El paladar, está formado por hueso en su porción
anterior, (paladar óseo), y por tejidos blandos en su parte
posterior, (paladar blando o velo del paladar). El hueso que
rodea el paladar óseo, donde están situados los
dientes, es el reborde alveolar.
Anatomía Básica: La nariz normal posee una
columela (porción blanda, central y anterior) recta apoyada
en un tabique central y recto. Los orificios nasales están
formados por los cartílagos alares que nacen como un arco
desde la columela y se apoyan en la base o ala nasal. Ellos son
los responsables de la armonía de la punta nasal.
Inmediatamente caudal se inicia el labio superior, que
presenta un músculo circular, el orbicular, que debe ser
íntegro. En el centro del labio, se sitúa el filtrum
(dos columnas y una depresión central) que terminan en el
arco de cupido (curvo como un corazón). El límite entre
el labio cutáneo y el rojo labial de la mucosa, está
dado por la línea blanca.
El paladar, está formado por hueso en su porción
anterior, (paladar óseo), y por tejidos blandos en su parte
posterior, (paladar blando o velo del paladar). El hueso que
rodea el paladar óseo, donde están situados los
dientes, es el reborde alveolar.
La parte anterior y central del paladar óseo, que
contiene a los incisivos, es llamada premaxila y se extiende
posteriormente hasta el forámen incisivo. La mayor parte del
paladar óseo se sitúa posterior al forámen
incisivo y está dado por los huesos maxilares y más
posterior, por los huesos palatinos.
El paladar blando, es muscular y móvil, está
implicado en la fonación y separa la cavidad oral de la
nasal. El músculo elevador del paladar y el tensor del
paladar forman una cinta que va de lado a lado y son los
encargados de mover el paladar.
Embriología y etiopatogenia:

Todas las estructuras situadas anteriores al foramen
incisivo (la punta nasal, el piso nasal, el labio, la encía,
el reborde alveolar y un pequeño segmento triangular del
paladar óseo) forman en su conjunto el paladar primario.
Posterior al foramen incisivo, (es decir en la mayor parte del
paladar óseo y en la totalidad del paladar blando), hablamos
de paladar secundario, que involucra a todo el paladar.
Así, al hablar de fisura del labio en realidad estamos
hablando de fisura del paladar primario, pues la fisura del labio
siempre conlleva anomalías de la nariz y del reborde
alveolar. Al hablar de fisura del paladar, en realidad estamos
hablando de la fisura del paladar secundario.
Si un mismo paciente presenta fisura del labio y del paladar,
estaremos frente a una fisura del paladar primario y
secundario.
Embriológicamente, la fisura del paladar primario se
explica por un defecto de fusión del mesénquima de dos
yemas laterales, que deben unirse a una yema central, para
originar así el labio, la punta nasal y el reborde alveolar.
Este defecto del avance del mesénquima puede ser unilateral
o bien bilateral, originando entonces la fisura unilateral o bien
la fisura bilateral. También, según el tamaño del
defecto de fusión de estas yemas, será la gravedad de
la fisura, que puede ir desde una simple muesca en el labio hasta
un amplio defecto que involucre todas las estructuras antes
mencionadas y comunique el piso de la fosa nasal con el
vestíbulo oral.
Embriológicamente, la fisura del Paladar Secundario se
explica por un defecto en la fusión de ambas conchas
palatinas. El embrión de 7 semanas presenta ambas conchas
palatinas en posición vertical al lado de la lengua. Antes de la semana 12,
la lengua desciende y las conchas palatinas ascienden y se
fusionan en la línea media para constituir el paladar. Este
movimiento arrastra
también a los músculos del paladar, que
deben fusionarse en la línea media para hacer la cinta
muscular del velo del paladar. Esta falla en la fusión de
los músculos palatinos será la responsable de los
problemas de lenguaje en estos
pacientes. También es importante que los músculos
palatinos se inserten normalmente en la Trompa de Eustaquio, pues
los RN con fisura palatina presentan déficit en la
aireación del oído medio y otitis secundarias a
ello.
También en este caso, según sea la magnitud del
error embriológico, podemos tener desde una simple
úvula bífida hasta un paladar óseo y blando
ampliamente separado, en toda su longitud y una gran
comunicación de la cavidad oro faríngea y la cavidad
nasal.
El error embriológico en el paladar primario y secundario
puede coexistir en un mismo paciente, de hecho la fisura más
frecuente es la del paladar primario y secundario unilateral
izquierdo. Embriológicamente nos explicamos esto, pues la
horizontalización de la concha palatina izquierda es
más tardía y por ello está más expuesta a
alterarse.
Cuidados generales y tratamiento del paciente
fisurado: Como hemos dicho anteriormente, el manejo de
estos pacientes, aunque exitoso en la actualidad, requiere de un
amplio y experimentado equipo multidisciplinario.
Idealmente, en un país como el nuestro, debiera existir
una centralización de todos
estos pacientes ojalá en un solo centro, para permitir tener
una experiencia nacional sólida y un gran número de
pacientes que permita evaluar los resultados y plantear
líneas de tratamiento.
Los cuidados de un paciente fisurado se inician desde su
nacimiento y en esta etapa son en general simples.
En el período de recién nacido nos preocupan cuatro
puntos: el diagnóstico y
pronóstico de la enfermedad, el consejo genético a los
padres y el estudio completo del paciente para descartar otras
anomalías (la mayoría de los casos no presenta otras
anomalías) pues hay casos en que la fisura forma parte de
síndromes malformativos.
El recién nacido fisurado no tiene problemas deglutorios
y de presentar trastornos para alimentarse, este es debido a una
succión deficitaria pues pierde presión, lo que no es un
problema frecuente. Posee los reflejos de succión normal. El
problema se soluciona al hacer llegar la leche a la cavidad oral
posterior, ya sea con un chupete largo y blando, o con un chupete
ancho que pueda ocluir su defecto palatino. Es mejor tener al
niño elevado 45 ° para evitar la regurgitación de
leche por la nariz. La madre debe ser involucrada desde un
comienzo y debe aprender los trucos para facilitar la alimentación del niño.
Los pacientes fisurados del paladar primario y/o secundario,
no tienen problemas respiratorios asociados. Solamente existe
problema respiratorio en el paciente portados del síndrome
de Pierre Robin. Este síndrome se caracteriza por un
recién nacido que presenta una mandíbula muy
pequeña (micrognatia) y/o muy posterior, (retrognatia)
asociada a una lengua grande (glosoptosis), que no tiene
suficiente espacio en la cavidad oral y cae hacía
atrás, produciendo una severa obstrucción respiratoria
de reposo y al dormir. En estos casos, dado que la
obstrucción es producto de la mandíbula y la lengua, y
no de la fisura palatina, el tratamiento consiste en poner al
niño en decúbito prono, a veces se fija la lengua al
piso de la boca y en los casos más severos se precisa
efectuar un cirugía precoz para elongar la mandíbula,
que hoy día se hace con una distracción ósea. La
traqueostomía ha sido la opción clásica de
tratamiento de los casos más severos. La distracción
ósea sería hoy la técnica de elección. En
aquellos centros donde la Cirugía Plástica está
bien desarrollada.
Cuando la fisura es del paladar secundario, el mal
funcionamiento de la trompa de Eustaquio impide la correcta
aireación del oído medio por lo que es importante la
prevención de patología del oído medio. La
prevención de otitis media en estos pacientes es efectuada
por el otorrinolaringólogo, quien efectúa una simple
operación, consistente en favorecer la aireación del
oído medio efectuando un orificio en el tímpano y
dejando un tubo para que este permanezca abierto y favorecer
así la aireación hacia el exterior (miringotomía y
tubos de ventilación o colleras). La prevención del
déficit auditivo de estos pacientes es muy importante para
la posterior adquisición del lenguaje y en el último
tiempo su reconocimiento ha
permitido disminuir de manera importante este Problema.
El Cirujano Plástico inicia el tratamiento del paciente
fisurado en la etapa de recién nacido y muchas veces termina
efectuando las últimas operaciones cuando su paciente es
adulto.
2.- Lactante menor:En esta etapa, y cerca de los
3 meses de edad se efectúa la reparación de la fisura
del paladar primario. Se opera el labio (queiloplastía o
plastía labial) cerrando el defecto cutáneo y haciendo
una reposición y cierre del músculo orbicular
defectuoso. Además en esta etapa se efectúa una
reparación de la nariz (nasoplastía) y se cierra la
porción más anterior del paladar óseo si éste
está abierto.
Algunas veces el ortodoncista efectúa ortopedia
prequirúrgica, es decir coloca unos aparatos intraorales o
externos, para acomodar los tejidos óseos y facilitar la
cirugía posterior.
3.- Lactante mayor:Cerca del año de
edad, se efectúa la reparación de la fisura del paladar
secundario, es decir se reparan el paladar óseo y el paladar
blando, (palatoplastía), con el fin de separar la cavidad
oral de la cavidad nasal y sobre todo, con el fin de proveer al
niño de una adecuada caja de resonancia y de un paladar
móvil para articular los fonemas y poseer así un
lenguaje normal. Idealmente el paladar debe estar reparado cuando
se inicia la adquisición del lenguaje, que es a partir de
los 12 meses de edad. Algunos grupos operan más precozmente
a estos niños, con el fin de
obtener un mejor desarrollo del lenguaje, sin embargo estas
operaciones muy precoces producen defectos en el crecimiento
facial y por ello este tema está en constante estudio y
discusión.
En términos generales, podemos decir que un paladar
cerrado bien, al año de edad, más o menos en el 70% de
los casos hablará bien y no necesitará de otros
tratamientos.
4.- Edad preescolar y
escolar: Todos los pacientes fisurados ya
deberían estar operados. Muchas veces hacemos retoques
estéticos de cicatrices y nos preocupamos de la
adquisición y desarrollo del lenguaje y del apoyo
psicológico. Además en esta etapa hacemos control por otorrinos, terapia de
lenguaje, fonoaudiología, ortodoncia, etc.

Manejo de secuelas y problemas de crecimiento
asociados: En el 30% de los pacientes, a pesar de
estar bien operados, se presentan secuelas con problemas
de voz gangosa (hipernasalidad) y con errores o imposibilidad de
articular fonemas y de tener una buena caja de resonancia, esto
es lo que conocemos como insuficiencia velo faringea. Tiene
tratamiento médico y también quirúrgico,
según sea el caso.
En un porcentaje variable de pacientes, según sea la
gravedad de la fisura que presentaron, el sitio donde fueron
tratados y la edad en que fueron
tratados, se presentan secuelas de crecimiento maxilar. Por este
motivo, en la adolescencia estos pacientes
pueden ser exitosamente operados de su maxilar y/o mandíbula
para tener un perfil armónico y una buena mordida.
También en la adolescencia, efectuamos
septorrinoplastías con fines estéticos y funcionales
(todos estos pacientes presentan grados variables de desviación
del tabique septal).
MALFORMACIONES
CRÁNEOFACIALES
Fisuras Faciales:

La fisura labio palatina es la fisura facial más
frecuente, por ello la describiremos en un punto aparte.
Las otras fisuras faciales, son mucho más raras y se
manifiestan con una hendidura en los tejidos blandos y en los
huesos del cráneo y de la cara. Según su intensidad
pueden ir desde una muesca en la comisura oral o en el
párpado, hasta una dramática deformidad que afecte toda
la cara y comunique la órbita con la nariz y la boca en un
gran defecto facial, como el observado en la figura 1 que
representa una fisura facial N° 3. Figura 1
Según la dirección que lleve esta fisura y tomando
como referencia la órbita y la boca, Tessier clasificó
estas fisuras, con números del 0 al 14. Los números del
0 al 7 representan las fisuras faciales y del 8 al 14, su
extensión craneal. Estas fisuras pueden ser únicas o
múltiples en un mismo paciente. (Figura 5).

El tratamiento de esta patología es muy complejo,
de una manera amplia, requiere tratar las partes blandas y el
esqueleto óseo así como los diferentes órganos
afectados por la fisura, por ello requiere de un equipo
multidisciplinario formado por varios especialistas.
Craneosinostosis
Craneosinostosis es el cierre prematuro de una (o varias)
suturas craneales. Cuando esto ocurre, el cráneo deja de
crecer en la zona sinostosada y compensatoriamente crece más
en las zonas donde las suturas aún no están osificadas,
para así poder acomodar el crecimiento del cerebro
subyacente. Se manifiesta clínicamente por una deformidad
craneal, de grado variable, según que suturas estén
alteradas. De una forma simple podemos decir que el aspecto de la
cabeza de un paciente con craneosinostosis presentaría una
región plana y otra abollonada. Su frecuencia es de 4 por
10.000 nacimientos. Su etiología es variable (genética, cromosómica,
metabólica etc.). La craneosinostosis puede ocurrir como un
hecho aislado o puede ser parte de un síndrome con otras
malformaciones asociadas. Si la sinostosis es múltiple y
severa, puede haber un impedimento al crecimiento del cerebro,
ocasionando desde hipertensión endocraneana
hasta microcefalia y déficit intelectual. Si la sinostosis
no es múltiple, el crecimiento cerebral es normal y no hay
déficit intelectual.
La craneosinostosis simple se debe a la fusión
prematura de una de las 6 suturas principales de la bóveda
craneal. También se las llama cráneo estenosis, pero
este término refleja más el resultado de la
craneosinostosis. Ocurren más frecuentemente que las formas
sindromáticas y a diferencia de éstas ocurren como un
hecho esporádico no ligado a la herencia. Según la
sutura fusionada, será la forma anómala que
presentará el cráneo y según sea esta forma, es
como llamamos a esta deformidad. Las diferentes craneosinostosis
simples son: (figura 6 y 7).


El tratamiento de las craneosinostosis simples lo realizan en
conjunto el Cirujano Plástico y el Neurocirujano. Es
bastante exitoso y se efectúa resecando la sutura fusionada
y remodelando los segmentos óseos deformes. Debe operarse
precozmente para evitar la restricción al crecimiento
cerebral y para aprovechar el potencial de crecimiento cerebral
(que está presente hasta los 3 años de edad) que es en
sí un factor remodelante de la bóveda craneal.
Las craneosinostosis sindromáticas corresponden a
un grupo de anomalías craneofaciales muy complejas. Las
manifestaciones clínicas derivan del efecto de la sinostosis
en múltiples suturas, de la bóveda craneal, de la base
del cráneo y de la cara. Además este grupo de pacientes
tiene anomalías en otras regiones distantes del esqueleto
craneofacial.
En general, presentan una forma de transmisión
hereditaria (autonómica recesiva o dominante), son muy
deformantes, difíciles de tratar y se alteran en grado
variable, desde el cráneo, las órbitas, la región
centro facial, el mecanismo de oclusión y masticación,
la visión, la vía aérea superior y el desarrollo
intelectual.
Al contrario de las craneosinostosis simples, en que cuando
uno corrige el área de sinostosis el crecimiento
craneofacial continúa normal, en las complejas, el
crecimiento del esqueleto craneofacial no sigue un patrón
normal por lo cual se requieren múltiples intervenciones a
través de la vida de estos pacientes. Por esto mismo, se
requiere de un amplio y experimentado equipo de
especialistas.
Las craneosinostosis sindromáticas más
representativas son:

Enfermedad de Crouzon: craneosinostosis múltiple
que origina un aspecto característico (frog- like facies)
con deformidad del cráneo, cara corta (maxilar
hipoplásico) ojos salidos y separados, (exoftalmos por
orbita pequeña e hiperteleorbitismo), estrabismo, mal
oclusión con protrusión de la mandíbula y
mentón pequeño.
Síndrome de Apert: enfermedad similar al Crouzon,
también con transmisión autonómica dominante, que
además de las deformidades craneales y faciales, presenta
sindactilia simétrica de ambas manos y pies y en ocasiones
fisura palatina, parálisis de músculos oculares y
ptosis palpebral.
Síndrome de Pfeiffer: similar a los anteriores por
el mecanismo de transmisión y por el aspecto facial, se le
distingue por presentar un pulgar y primer ortejo mas largo.
Síndrome de Saethre-Chotzen: similar a los
anteriores, se le distingue por presentar además
braquidactilia.

Disostosis
Craneofaciales

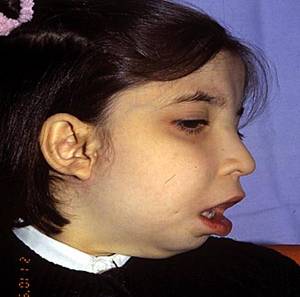
Microsomía hemifacial: Es una malformación
presente al nacimiento, no es tan rara (1/ 3.000 RN vivos). Se
manifiesta por hipoplasia (en grado variable) de la
hemimandíbula y del maxilar, que además presenta
malformación auricular en ese lado.
Es generalmente unilateral, aunque raramente puede ser
bilateral. No se conoce bien su etiología, pero se supone
que se originaría in útero, por una alteración
vascular en la arteria estapedia (arteria embrionaria del 1er y
2do arco branquial y precursora del sistema carotídeo).
Clínicamente observamos un paciente con asimetría
facial, con su mandíbula y mentón desplazados hacia el
lado afectado (laterognatia debido a hipoplasia del ramo maxilar)
con alteraciones de la mordida, con una malformación
auricular variable, desde papilomas preauriculares hasta microtia
(hipoplasia del pabellón auricular y del CAE) o anotia.
También puede tener parálisis facial congénita a
ese lado y diversa alteración de los músculos
masticatorios, por eso al intentar abrir la boca, exagera su
deformidad.
Antiguamente se la conocía como Síndrome del Primer
y Segundo Arco Branquial, pues todas las estructuras que derivan
de estos arcos, están alteradas en grado variable.
Su tratamiento comprende la reparación de los tejidos
óseos y blandos alterados. El tratamiento se inicia en la
primera infancia con reparación
del nervio facial y elongación del ramo mandibular
(distracción ósea). A los 6 años de edad se
efectúa la reconstrucción auricular (con
cartílagos costales) y en la juventud se efectúa
cirugía del maxilar y la mandíbula (osteotomías e
injertos óseos).
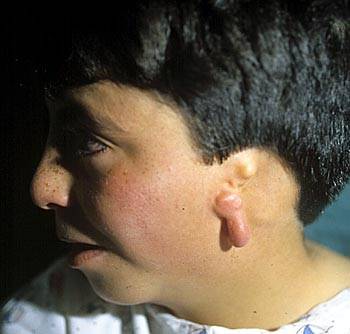
Síndrome de Treacher Collins: También
llamado Disostosis Mandibulofacial. Clínicamente podría
tener un aspecto parecido a la microsomía hemifacial, pero
este síndrome es siempre bilateral y además no se
presenta como casos aislados debidos a un accidente vascular in
útero, sino que tiene un patrón de trasmisión
genética autonómico dominante. Según Tessier, este
síndrome correspondería a la combinación de las
fisuras faciales (6,7 y 8) de la región
órbito-malar.
Anatómicamente se presenta como una hipoplasia con
ausencia de hueso (fisura) en el zigoma (o malar) y en la
órbita. Además existe una mandíbula con un ramo
hipoplásico, malformación auricular, inclinación
antimongoloide de los párpados y colobomas (detecto en el
borde libre del párpado).
El tratamiento de estos pacientes se inicia en la infancia y
comprende la reconstrucción de partes blandas (orejas y
párpados) así como injertos óseos vascularizados
en el área del malar y cirugías en maxilar y
mandíbula. Figura 6.
Figura 6
MALFORMACIONES ORBITARIAS
La forma y la posición de las órbitas en la cara,
son importantes desde un punto de vista funcional (visión
binocular) y también estético. La mal posición de
la órbita en el eje transversal de la cara, traducido por
ojos muy separados o muy juntos, es lo que llamamos hiper o
hipotelorismo. Cuando una órbita está mal posicionada
en sentido vertical, más alta o más baja, hablamos de
distopias orbitarias.

Hipertelorismo e Hipotelorismo: El hipertelorismo es un
aumento de la distancia de las paredes internas de la
órbita, que en el adulto normal corresponde a 23-28 mm de
distancia interorbitaria (medición efectuada en una
Rx) y de 30-35 mm de distancia intercantal (medición
efectuada en el paciente, tomando como referencia ambos cantos
internos). El hipertelorismo no es en sí un
diagnóstico, sino es la manifestación clínica de
otra patología como podría ser una fisura facial de la
línea media (0-14) o una craneoestenosis o un tumor.
El hipotelorismo es una menor distancia entre ambas
órbitas.
En ambos casos el tratamiento es la cirugía ósea con
movilización de las órbitas hacia la línea media,
resecando un segmento óseo en la región de la frente y
la nariz y además tratando la enfermedad de base… Es una
patología de resorte multidisciplinario en que intervienen
el Cirujano Plástico, el Neurocirujano y el
Oftalmólogo.
Distopia Orbitaria: Corresponde a una mal posición
orbitaria. Es posible corregirla quirúrgicamente,
movilizando la órbita y su contenido, como un cubo,
efectuando osteotomías para resecar un segmento óseo en
caso de necesitar elevarla) o resecar un segmento de maxilar y
zigoma, en caso de necesitar descenderla
Malformaciones
del maxilar y la mandíbula
El maxilar y la mandíbula son muy importantes en el
aspecto estético del tercio inferior de la cara y
también en el aspecto funcional de la forma de morder y de
las relaciones que presentan los dientes entre sí
(oclusión dental normal y patológica).
Para poseer una buena estética y una mordida normal, el
maxilar y la mandíbula deben relacionarse
armónicamente, en forma, tamaño y posición.

El estudio de las relaciones óseas del maxilar y la
mandíbula se hace por medio del examen clínico, por
medio del estudio de la oclusión dental (moldes de yeso) y
por medio del estudio radiológico cefalométrico, que
mide los ángulos en que se relacionan el maxilar y la
mandíbula entre sí y con las otras estructuras
faciales. Figura 7.
En el tratamiento de esta patología se requiere de la
estrecha relación entre el cirujano plástico,
maxilofacial y el ortodoncista. Ninguno de estos especialistas
debe intentar solucionar por sí sólo el problema, ya
que al tratar de modificar la posición dental (tratamiento
ortodóncico exclusivo) sin corregir la deformidad del
esqueleto facial, tendremos perfiles disarmónicos y
recidivas de la mal oclusión. De la misma forma, el cirujano
plástico debe prever la forma en que encajarán los
dientes después de una osteotomía de maxilar y
mandíbula y preparar esta mordida antes y después de su
cirugía para obtener óptimos resultados.
La relación oclusal normal es llamada ANGLE
I .El primer molar superior encaja justo con el primer
molar inferior. La relación ósea del maxilar y la
mandíbula (el maxilar discretamente delante de la
mandíbula) es armónica y eso se traduce en un perfil
normal. Figura 8-A.
La mal oclusión tipo ANGLE II es aquella en
que los dientes de la arcada superior y el maxilar se sitúan
más anteriores que los dientes de la arcada inferior. Esto
puede ser secundario a un maxilar muy grande y adelantado y/o a
una mandíbula muy pequeña y retraída. Se
manifiesta clínicamente por un perfil convexo o cara de
pájaro.
El tratamiento es la cirugía ortognática, efectuando
un avance de la mandíbula, una reproposición del
maxilar o una combinación de ambas cosas.
La mal oclusión tipo ANGLE III es aquella
en que los dientes de la arcada inferior y la mandíbula, se
encuentran por delante de los dientes superiores y del maxilar.
Esto ocasiona un perfil cóncavo, con protrusión
exagerada de la mandíbula (Prognatismo).


Figura 8
Definiremos someramente las malformaciones del |
| Retrognatia: Proyección o Prognatismo: Proyección y tamaño |
Microgenia y Macrogenia: Se refiere al tamaño y
posición del mentón, únicamente, por lo cual no
influye en la oclusión y puede asociarse a cualquier tipo
oclusal, normal o patológico. El tratamiento quirúrgico
consiste en una osteotomía de reposicionamiento y
remodelación del mentón y en algunos casos mínimos
de microgenia se puede colocar una prótesis mentoniana con fines
netamente estéticos.
Laterognatia: Corresponde a una
asimetría con desviación mandibular y puede ser
secundaria a malformaciones craneofaciales, a traumatismos, a
tumores etc. etc. También se corrige quirúrgicamente,
según su etiología y magnitud.
Hipoplasia maxilar: Es un maxilar
superior poco desarrollado en el sentido antero posterior
(retrusión maxilar) o en el sentido vertical (colapso
maxilar vertical). La causa más frecuente de hipoplasia
maxilar es la fisura labio palatina, cuya secuela es un defecto
del crecimiento óseo maxilar.

Hipertrofia maxilar: Es un desarrollo exagerado
del maxilar superior. Cuando es en sentido vertical, ocasiona la
Sonrisa Gingival, que es la exposición exagerada de
encía. Cuando la hipertrofia es en sentido antero posterior,
ocasiona el perfil convexo con dientes salientes y mal
oclusión Angle II. Se corrige con cirugía ósea
maxilar.
Síndrome de Pierre Robin: Es una
micrognatia y retrognatia congénita severa, de carácter
esporádico (no hereditaria) que se asocia a fisura del
paladar y a una lengua grande, por lo cual estos recién
nacidos presentan problemas respiratorios por obstrucción de
la vía aérea superior. Ver manejo general y de
problemas respiratorios en la sección de Fisuras del Labio y
Paladar, manejo del recién nacido fisurado.
V. CONCLUSIONES
·
La mayoría de las malformaciones de la cara y el cráneo
se debe a factores congénitos, pero no debemos de olvidar
que un gran número de estas se dan por una causa
multifactorial.
VI. BIBLIOGRAFÍA
·
Abraham Abramovich. "Embriología de la
región maxilofacial". Tercera edición. 1997. Editorial
Médica Panamericana.
·
Guillermo Raspall." Cirugía Maxilofacial. Patología
quirúrgica de la cara, boca, cabeza y cuello."Editorial
Médica Panamericana, 1997.
·
Lagman. "Embriología Médica".10a
edición. Editorial Panamericana. Buenos Aries- 2007.
·
Moore Persaud. Embriología clínica.7ª
edición. Editorial el sevier España S.A-2004.
Autor:
Luis Alberto Llanos Vera
CURSO: EMBRIOLOGIA MéDICA
DOCENTES:
DR. RODIL CRUZALEGUI HENRIQUEZ
DR. LUIS FLORIAN ZAVALETA
DR. JAVIER ALVAREZ
DR. GUILLERMO FONSECA
UNIVERSIDAD NACIONAL DE TRUJILLO
FACULTAD DE MEDICINA
ESCUELA DE MEDICINA HUMANA
DEPARTAMENTODE MORFOLOGÃŒA HUMANA
TRUJILLO - 2008
 Página anterior Página anterior | Volver al principio del trabajo | Página siguiente  |