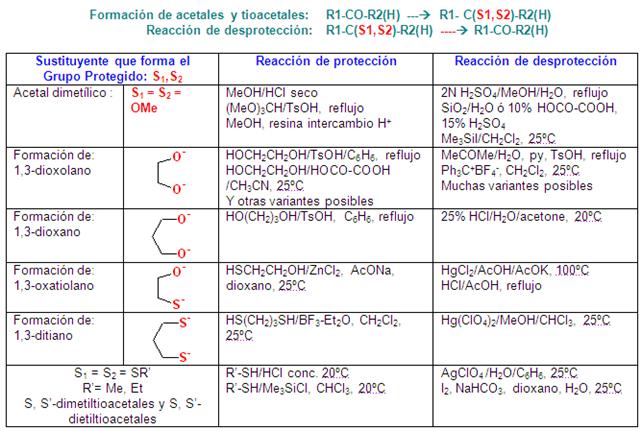
Protección
de grupos carbonilo: R1-CO-R2
Con mucha frecuencia, se tiene que proteger
s los grupos carbonilo de aldehídos y cetonas
alifáticas y/o aromáticas de la acción
de:
Nucleófilos fuertes, incluidos
los organometálicos.Agentes reductote: ácidos,
básicos e hidrurosAlgunos oxidantes
* El diazometano CH2N2, se genera a partir
de Diazald
(N-metil-N-nitroso-p-toluensulfonamida)
Protección
de tioles: RSH
En la síntesis de péptidos y
proteínas se halla involucrada el aminoácido
cisterna, igualmente en la síntesis de varios
fármacos se hallan funciones tío o sulfuro formando
heterociclos muy singulares que requieren muchas veces de ser
protegidos. Esta protección se lo hace
transformándolos temporalmente en tioéteres o
tioésteres.

PROTECCIÓN DE ALDEHÍDOS Y
CETONAS
Una forma de protección de las
cetonas y los aldehídos es su conversión en
acetales.
Los acetales se pueden desproteger en La protección de la cetona en | hidroxilo. |

En el siguiente esquema se da la secuencia
de síntesis completa que permite conseguir la
reducción del éster sin afectar a la
cetona:

En la primera etapa la cetona se convierte
en un acetal cíclico por reacción con etilenglicol
en presencia de un catalizador ácido. En la segunda etapa
se reduce el éster con LiAlH4. Este reactivo no ataca al
acetal. Finalmente, en la tercera etapa el alcohol-acetal se
trata en medio ácido acuoso. En estas condiciones el
acetal resulta hidrolizado regenerándose el grupo
carbonilo cetónico. Cada una de las tres etapas es
quimioselectiva puesto que en cada una de ellas se consigue la
reacción preferente de un grupo funcional en presencia de
otro.
PROTECCIÓN DE ÁCIDOS
CARBOXÍLICOS
a) ésteres de etilo y
metilo
La forma de protección Los ésteres más |
|

La desprotección se lleva a |
|

b) ésteres de bencilo
La desprotección de ésteres
de etilo o metilo puede ser problemática en sistemas
polifuncionales debido a la elevada acidez o basicidad que se
debe emplear en el proceso de hidrólisis. Por ello se
emplean otro tipo de ésteres que permiten efectuar la
etapa de desprotección en condiciones neutras o de baja
acidez.
Los ésteres de bencilo se |
| |

c) ésteres de t-butilo
Los ésteres de t-butilo se |
| |

PROTECCIÓN DE ALCOHOLES
a) Como acetales
El DHP (dihidropirano) se emplea para |
|

b) Como éteres de bencilo
Puesto que los éteres son uno de los
grupos funcionales menos reactivos no es de extrañar que
muchos de ellos se empleen como grupos protectores. Sin embargo,
la inercia química de los éteres es un
inconveniente a la hora de utilizarlos como grupos protectores
porque la etapa de desprotección obliga, en muchos casos a
la utilización de condiciones de reacción muy
drásticas.
Es por ello que, en la Uno de los éteres más |
| |

La etapa de protección se consigue
por la ionización previa del alcohol, por ejemplo con NaH,
seguida de ataque SN2 del alcóxido generado sobre bromuro
o cloruro de bencilo.
Los bencil éteres son muy |
| |

c) Como tritil éteres
Los tritil éteres, o éteres
de trifenil metano, se emplean para la protección
quimioselectiva de hidroxilos primarios. Los grupos hidroxilo
secundario y terciario, al estar estéricamente más
impedidos que los primarios, no forman éteres de tritilo
porque el cloruro de trifenilmetilo (cloruro de tritilo) es un
reactivo muy voluminoso.
Los éteres de trifenilmetano |
| |

La desprotección de este tipo |
|

c) como silil éteres
Los silil éteres se obtienen |
| |



Los silil éteres se pueden Esta desprotección se basa en |
| |

El otro producto de esta reacción es
una sal del anión alcóxido (RO-M+). Para obtener el
alcohol se procede a efectuar una etapa de hidrólisis para
provocar la protonación del anión
alcóxido.
El tamaño de los tres reactivos de
sililación , aumenta en el siguiente orden:
![]()
Aumenta el tamaño del reactivo de
sililación
d) protección como
ésteres
Los alcoholes también se pueden
proteger mediante su conversión en
ésteres.
Uno de los ésteres más |
| ||

PROTECCIÓN DE AMINAS
El par electrónico libre situado
sobre el átomo de nitrógeno de las aminas es el
responsable de la nucleofilia y de la basicidad de éstas.
La forma evidente de ocultar las propiedades básicas y
nucleofílicas de las aminas es su conversión en
compuestos en los que el par electrónico del
nitrógeno esté conjugado con un grupo
electrón-atrayente.
La conversión de aminas en |
| |

Esta protección tiene un
inconveniente: la etapa de desprotección. Las amidas son
poco reactivas y la hidrólisis del grupo amida hay que
efectuarla en condiciones de alta basicidad (o acidez) y
temperatura que puede afectar a otros grupos funcionales
presentes en la estructura. Por ello, las aminas se suelen
proteger en forma de uretanos y no de amidas.
En los uretanos la densidad
electrónica del átomo de nitrógeno
también está disminuida por conjugación con
un grupo carbonilo. La ventaja de estos protectores es que pueden
eliminarse en condiciones suaves y muy
quimioselectivas.
Uno de los reactivos empleados en la
protección de aminas en forma de uretanos es el cloruro de
t-butiloxicarbonilo. Los uretanos obtenidos con este reactivo se
abrevian como RNHBoc

La reacción de los RNHBoc con
ácidos acuosos, en condiciones suaves de acidez y
temperatura, genera un ácido carbámico que es
inestable y se descarboxila in situ dando lugar a la
amina libre. Otro tipo de uretanos empleados en la
protección de aminas son los que se obtienen en la
reacción con cloruro de benciloxicarbonilo.
Las aminas (RNH2) protegidas como uretanos
de benciloxicarbonilo se abrevian como RNHCBz

Estos uretanos se desprotegen en
condiciones neutras mediante una reacción de
hidrogenólisis.
Desprotección de N-CBz
derivados:
1º. Generación del |
| |

2º. Descarboxilación |
|

ACTIVACIÓN
DESACTIVACIÓN
Muchas veces es necesario activar una parte
de una molécula para que la reacción ocurra
preferentemente en ese lugar o sencillamente es necesario
desactivar un grupo activante, para disminuir la reactividad de
la molécula. Esto puede observarse en los siguientes
ejemplos.
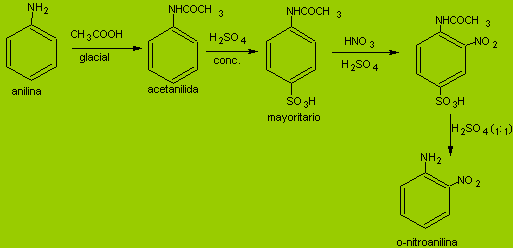
Cuando se pretende obtener la orto
nitroanilina a partir de la anilina, una nitración directa
de la misma, proporcionaría una mezcla de isómeros
orto y para anilinas, debido a que el grupo –NH2 es
activante de la molécula de benceno para las sustituciones
electrofílicas.
De manera que la estrategia debe contemplar
una necesaria disminución del poder activante del grupo
amino, lo que se consigue transformándolo en un grupo
amida, con la ácido acético o anhídrido
acético.. Se forma la acetanilida.
El nuevo grupo sigue siendo orientador orto
para, preferentemente para, debido a que por ser voluminoso el
grupo amida, se origina un efecto estérico, que impide a
que cualquier electrófilo pueda aproximarse exitosamente a
la posición orto del anillo bencénico, estando
libre la posición para.
Esto se puede aprovechar para sulfonar la
acetanilida, obtener el isómero mayoritario
para-sulfo-acetanilida, que luego es sometida a nitración,
que luego por hidrólisis ácida del grupo amida y
del grupo sulfónico, se libera la orto nitro anilina
solicitada.
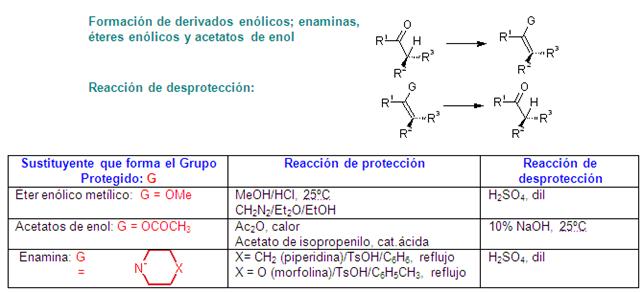
Frecuentemente se requiere que los carbono
alfa de un compuesto carbonílico, por ejemplo una cetona,
se transformen en buen nucleófilo, para participar de
reacciones con altos rendimientos, esto se puede lograr de las
dos maneras siguientes:
|
|


SÍNTESIS DE MOLÉCULAS
POLIFUNCIONALES
Para la síntesis de moléculas
polifuncionales, es necesario la aplicación de los
principios generales y fundamentalmente los tres principios que
se acaban de explicar (simetría, selectividad y control),
de acuerdo a las características la molécula
objetivo que se desee sintetizar.
MOb. 07. Proponer un plan de |
| |

Solución. LA MOb 7 es
bifuncional y los grupos son hidroxílicos: uno terciario y
otro primario. El esqueleto carbonado presenta dos
ramificaciones: un metilo y un etilo. Puesto que la
molécula posee poca simetría, es improbable que
ambos grupos puedan introducirse
simultáneamente.
Para empezar por el final,
considérese cómo se podría intentar la
introducción de una función, en presencia de la
otra. En forma arbitraria, inténtese introducir un grupo
oxhidrilo primario en una molécula precursora que tenga el
OH terciario. Para esto se tiene dos posibilidades:
Hidroboración/oxidación de una olefina terminal y
adición de un reactivo de Grignard al
formaldehído.
Si se analiza con detenimiento, la segunda
ruta es improbable, puesto que es imposible formar un compuesto
de Grignard del mismo ya que el OH terciario es suficientemente
ácido como para descomponer el Grignard que estuviera
formándose. La reacción de
hidroboración/oxidación del primer camino resulta
ser un proceso selectivo: no afecta el grupo hidroxilo ya
existente en una molécula
| Se debe considerar ahora cómo Puesto que este alcohol | |

Los alcoholes terciarios se forman
comúnmente por adición de un reactivo de Grignard
una cetona y el doble enlace no altera la reacción, siendo
por lo tanto también selectiva. De este modo, puede
prepararse el alcohol partiendo de un precursor cetónico
no saturado.
| ||||

Ahora se puede centrar la atención
en la construcción del esqueleto carbonado. Se puede
recurrir para ello nuevamente a una síntesis de Grignard;
sin embargo se puede observar que el último precursor
está estructuralmente relacionado con el éster
acetoacético ya que la estructura típicamente se
puede obtener a partir del éster acetoacético o es
una metil cetona o es un producto de transformación de
ella.

En consecuencia, se puede obtener el
compuesto intermedio por alquilización del acetoacetato de
etilo: primero con bromuro de etilo y luego con bromuro de alilo,
luego, se hidroliza y descarboxila el producto, para generar la
MOb 7.
El bromuro de alilo se prepara de la |
| |

MOb 08. Las feromonas, son agentes |
| |

Solución:
El grupo funcional de la Con estas consideraciones se puede | a)
b)
|

Por otra parte, el isómero
geométrico epóxido tiene que obtenerse de su
similar alqueno cis. Entonces el alqueno precursor debe
ser:

La deshidratación de un El plan de síntesis que se Los compuestos (A) y (B), deben ser Las aplicaciones en los siguientes ¿cómo se justifica la ¿Cómo se sintetiza la En la solución de ellos se |
| |

MOb 09.
(1E,4E)-hepta-1,4-dienilcyclopentano:
Proponer un plan de síntesis, a
partir de materiales simples y los reactivos necesarios, para la
siguiente molécula:
Solución.
Una adecuada combinación de la
síntesis acetilénica y de Grignard, permite
elaborar un plan de síntesis factible a partir del
ciclopentanol y el acetaldehído. Es necesario hacer notar
que a esta altura de los conocimientos de las reacciones, no
permite aún plantearse la preparación del anillo de
cinco miembros.
Es necesario tener cuidado que el haluro
que reacciona con el ión acetiluro deba ser necesariamente
primario, con los haluros secundarios y terciarios, la
reacción que predomina es la de eliminación y no
ocurre la sustitución.
Se aprovecha adecuadamente la
bromación del sistema alílico que se forma con la
succinimida,
Los acetiluros también son
nucleófilos que pueden atacar un epóxido formando
luego de la hidrólisis ácida un alcohol.

MOb. 10.
¿Cómo se efectúa |
|

Solución:
La MOb 10, incrementa en un grupo metilo la
cadena alquílica del compuesto aromático. El punto
de instauración sugiere que el mismo puede prepararse a
partir de la deshidratación de un alcohol. Esto permite
pensar que el grupo metilo proviene de un compuesto de Grignard,
que a su vez forma simultáneamente el alcohol.
Esta reacción sólo El alcohol requerido puede formarse |
| ||

Autor:
Wilbert Rivera Munoz
[1] Borrell J.I. “Análisis
Retrosintético”. Institut Quimic de Sarriá,
Universitat Ramon Llull. Grup d’Enginyeria Molecular
(GEM).
 Página anterior Página anterior | Volver al principio del trabajo | Página siguiente  |